Three human diseases, dyskeratosis congenita (DKC), aplastic anemia (AA), and idiopathic pulmonary fibrosis (IPF), have been linked to mutations within the genes that encode for the two telomerase essential core components, telomerase RNA (TR) and telomerase reverse transcriptase (TERT); telomerase-associated proteins, DKC1 encodes for dyskerin and Nola3 encodes for Nop10; and recently to one of the six proteins that for the shelterin complex, TINF2 encodes for TIN2. Heterogeneous mutations show that even half the dose of telomerase is insufficient to maintain telomere length, resulting in erosion and loss of function, senescence, and apoptosis. The maintenance of telomere length in highly prolific cells, germline and stem cells, is crucial for the preservation of high populations and human health. In general point mutations, which leads to a single amino acid substitution, are better tolerated than frame shift and splicing junction mutations, limiting but not abolishing telomerase activity. The toleration of reduction and loss of telomerase function decreases with each subsequent generation. This anticipation, or progression of symptoms within the next generation, is characteristic of telomerase-deficiency diseases. The telomeres of the parental generation erode and when passed to their offspring begin this generation with shorter telomeres. The increase in severity of symptoms is linked with the progressive decrease of telomere length.
Dyskeratosis congenita (DKC) is an inherited disorder with clinical manifestations of skin hyperpigmentation (dark patches of skin), oral leukoplakia (white spots inside the mouth), and nail dystrophy (lack of nails). The majority of deaths occur from bone marrow failure, immunodeficiency, pulmonary complications, and malignancies. The X-linked recessive form has severe clinical presentations and caused by mutations found within the DKC1 gene that encodes for the dyskerin protein. The autosomal dominant form of the disease has been shown to be caused by mutations within the genes that encode TR as well as TERT. The autosomal recessive form of the disease has been shown to be caused by a mutation within Nola3, the gene that codes for Nop10. These mutations cause a reduction of telomerase activity leading to a limitation in stem cell capacity for proliferation. This reduction in proliferative capacity for high turnover cells leads in low counts for blood and immune cells, resulting in aplastic anemia. Bone marrow failure, brought on by the aplastic anemia, is the most common cause of death for patients with DKC. Families with this disease show anticipation, or the worsening of symptoms in subsequent generations because each generation begins with shorter telomeres than the previous Vulliamy et al, 2006.
Aplastic anemia (AA) is characteristically an acquired disease, however, there are rare constitutional forms of the disease that are found in patients with a strong familial history of various blood diseases. The disease has is connected with mutations with the genes that encode TR and TERT. This constitutional form of bone marrow failure is defined by low peripheral blood cell counts, hypocellular bone marrow, does not respond to immunosuppressive therapy, and included typical physical anomalies. The constitutional has associated with patients with DKC, however, there are cases lacking symptoms of DKC. The most common cause of death is due to bone marrow failure Fogarty et al, 2003.
Idiopathic pulmonary fibrosis (IPF) is a specific form of plumonary fibrosis with unknown cause, Plumonary fibrosis involves fibrotic lesion and scarring of the lung. The build up of excess scar tissue in the lungs results in reduced lung volume. The symptoms that typify the disease are chronic cough and shortness of breath. Some familiar types of IPF are caused by mutations in the genes that encode TR and TERT Armanios et al, 2007.
A standard nomenclature is used throughout the online database for consistency and clarity. Mutations designated according to the guidelines and recommendations for mutation nomenclature from the Human Genome Variation Society (HGVS). The nucleotides are numbered according to the coding DNA reference sequence, intronic positions are numbered as an addition to the last nucleotide from the preceding exon or as a difference from the first nucleotide from the proceeding exon. The (?) indicates uncertainty in the description of the mutation while frame shifts are described by the first affected amino acid and from this position the number of amino acids to the stop codon.
| TR | TERC | TER | TER1 | TLC1 | |||
|---|---|---|---|---|---|---|---|
| TERT | TRT | Est2 | p123 | TP2 | TCS1 | ||
| Gar1 | Nola1 | ||||||
| NHP2 | Nola2 | L7Ae | |||||
| Nop10 | Nola3 | ||||||
| dyskerin | Nola4 | Cbf5 | DKC1 | NAP57 | |||
| DnaK | hsp70 | ||||||
| Est1A | Smg6 | ||||||
| Est1B | Smg5 | ||||||
| TRF1 | TERF1 | TRBF1 | TRF | PIN2 | FLJ41416 | ||
| TRF2 | TERF2 | TRBF2 | |||||
| Pot1 | TEBPα | ||||||
| TINF2 | TIN2 | ||||||
| Rap1 | TERF2IP | DRIP5 | |||||
| TPP1 | TEBPβ | CLN2 | GIG1 | LPIC | |||
| MDV | GaHV (Gallid herpesvirus) | ||||||
Listed in gold is the nomenclature in use within the database with other common names and homologs to the right.
TR
TR mutations (autosomal dominant DKC, AA, IPF)
Numerous mutations causing nucleotide substitution, additions, and deletions have been documented within TR, the RNA component of the telomerase ribonucleoprotein (RNP), that have been connected with human diseases. TR contains the template that encodes for telomeric repeats and binds to telomerase reverse transcriptase (TERT) for DNA synthesis. Three domains characterize TR, pseudoknot that includes the template, conserved regions 4 and 5 (CR4-CR5), and ScaRNA domain for nuclear recruitment.
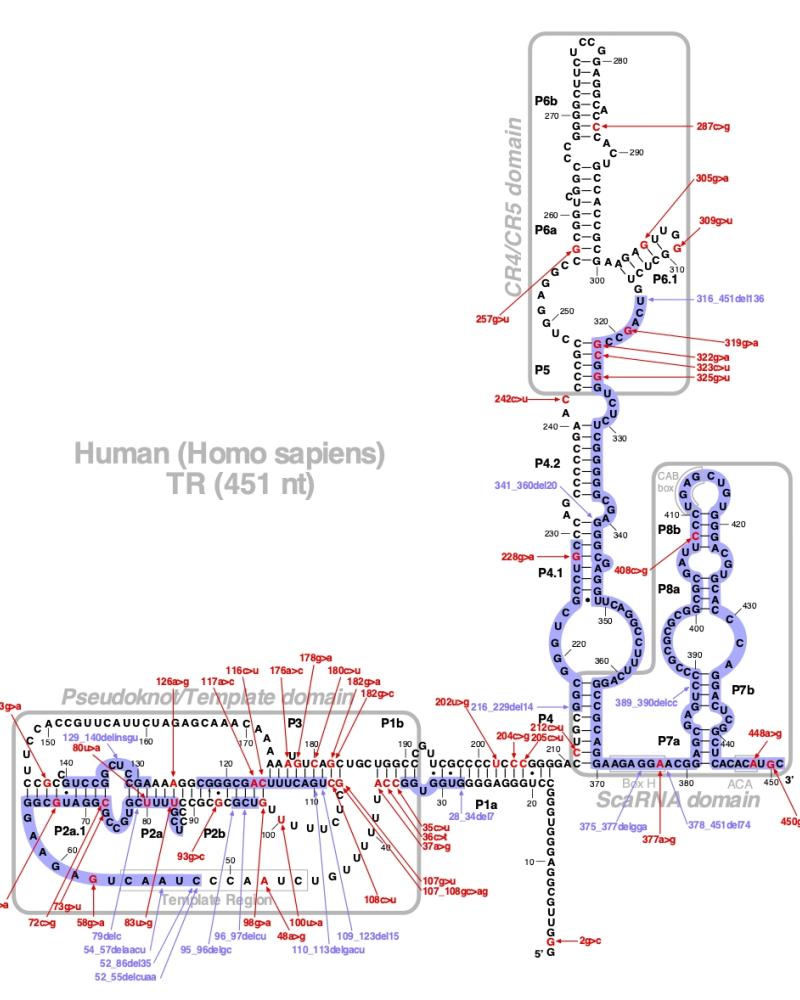
The above secondary structure for the 451 nt RNA component of the telomerase ribonucleoprotein (RNP) has indicated the location of mutations known to cause human diseases. The primary nucleotide sequence is in black, point mutations are colored red, while deletion mutations are shaded blue, and both are labeled. The Genbank accession number used is NR_001566 for the RNA sequence. Below is a description of the locations within the nucleotide sequence, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the RNA component of telomerase. The mutations are organized by domain from the 5'-terminus to the 3'-terminus.
TR mutations
| Domains | Mutation | Region | Presentation | References |
|---|---|---|---|---|
| Promoter | c.1-22C>U | Pulmonary Fibrosis | Justet et al, 2021 | |
| c.-771A>G | n/a | myelodysplastic syndrome | Carroll et al, 2011 | |
| c.-714insC | n/a | myelodysplastic sydrome | Carroll et al, 2011 | |
| c.-240delCT | n/a | myelodysplastic syndrome | Field et al, 2006 | |
| c.-99C>G | n/a | paroxysmal nocturnal hemoglobinuria, menorrhagia, anemia, and thrombocytopenia | Keith et al, 2004 Ortmann et al, 2006 |
|
| Pseudoknot | r.1-22C>U | Pulmonary Fibrosis | Borie et al, 2015 | |
| r.2g>c | 5' end | aplastic anemia | Marrone et al, 2007.1 | |
| r.17G>A | 5' end | Myelodysplastic syndrome / Acute myeloid leukemia | Schratz et al, 2020 | |
| r.23 G>C | Pulmonary Fibrosis | Justet et al, 2021 | ||
| r.28_34del7 | P1a | aplastic anemia | Xin et al, 2007 | |
| r.30G>A | Pulmonary Fibrosis | Justet et al, 2021 | ||
| r.35C>U | NA | Pulmonary Fibrosis | Justet et al, 2021 | |
| r.35c>u | P1b | hypoplastic myelodysplastic syndrome | Du et al, 2009 | |
| r.36c>u | P1b | aplastic anemia | Vulliamy et al, 2011 | |
| r.37a>g | P1b | autosomal dominant dyskeratosis congenita, idiopathic pulmonary fibrosis, dyspnea, aplastic anemia, and hepatic cirrhosis | Ly et al, 2005.2 Tsakiri et al, 2007 Calado et al, 2011 Han et al, 2009 |
|
| r.48a>g | Template | dyskeratosis congenita | Vulliamy et al, 2006 | |
| r.52_55delcuaa | Template | myelodysplasia and dyskeratosis congenita | Vulliamy et al, 2006 | |
| r.52_86del35 | Template | pulmonary fibrosis | Marrone et al, 2007.1 | |
| r.54_57delaacu | Template | dyskeratosis congenita | Jongmans et al, 2012 | |
| r.58g>a | template flanking | polymorphism | Vulliamy et al, 2002 Yamaguchi et al, 2003 Marrone et al, 2004 |
|
| r.67g>a | P2a.1 | aplastic anemia | Vulliamy et al, 2011 | |
| r.72c>g | P2a.1 | aplastic anemia | Vulliamy et al, 2002 | |
| r.73g>u | P2a.1 | dyskeratosis congenita | Yamaguchi et al, 2015 | |
| r.79delc | P2a | aplastic anemia | Vulliamy et al, 2006 | |
| r.80u>a | P2a | idiopathic pulmonary fibrosis | Stanley et al, 2015 | |
| r.83u>g | P2a | aplastic anemia, myelodysplastic syndrome | Vulliamy et al, 2011 | |
| r.91G>C | Pulmonary Fibrosis | Justet et al, 2021 | ||
| r.93g>c | P2b | dyskeratosis congenita | Collopy et al, 2015 | |
| r.95_96delgc | P3 | dyskeratosis congenita | Vulliamy et al, 2011 | |
| r.96_97delcu | P2b | autosomal dominant dyskeratosis congenita | Vulliamy et al, 2004 | |
| r.98g>a | P2b | idiopathic pulmonary fibrosis | Armanios et al, 2007 | |
| r.100u>a | P2b flanking | dyskeratosis congenita | Du et al, 2009 | |
| r.107g>u | P3 | aplastic anemia | Vulliamy et al, 2011 | |
| r.107_108gc>ag | P3 | autosomal dominant dyskeratosis congenita | Vulliamy et al, 2001 | |
| r.108c>u | P3 | idiopathic pulmonary fibrosis | Dai et al, 2014 | |
| r. 109_123del15 | P3_P2b | pancytopenia | Calado et al, 2009.2 | |
| r.110_113delGACU | Pulmonary Fibrosis | Justet et al, 2021 | ||
| r.110_113delgacu | P3 | aplastic anemia, myelodysplasia and leukemia | Vulliamy et al, 2002 Marrone et al, 2007.1 |
|
| r.116c>u | P2b | aplastic anemia and thrombocytopenia | Fogarty et al, 2003 Ortmann et al, 2006 |
|
| r.117a>c | P2b | aplastic anemia | Ly et al, 2005.1 | |
| r.126a>g | P2a | aplastic anemia | Vulliamy et al, 2011 | |
| r.129_140delinsgu | P2a | dyskeratosis congenita | Collopy et al, 2015 | |
| r.135G>C | Pulmonary Fibrosis | Justet et al, 2021 Borie et al, 2015 |
||
| r.143g>a | P2a.1 | autosomal dominant dyskeratosis congenita and aplastic anemia | Vulliamy et al, 2004 Parry et al, 2011 |
|
| r.164A>C | NA | Pulmonary Fibrosis | Justet et al, 2021 | |
| r.170C>A | NA | Pulmonary Fibrosis | Justet et al, 2021 | |
| r.176a>c | P3 | aplastic anemia | Vulliamy et al, 2011 | |
| r.178g>a | P3 | aplastic anemia | Marrone et al, 2007.1 | |
| r.180c>u | P3 | aplastic anemia | Marrone et al, 2007.1 | |
| r.182delG | Pulmonary Fibrosis | Justet et al, 2021 Borie et al, 2016 |
||
| r.182g>c | P3 | pleuroparenchymal fibroelastosis | Newton et al, 2016 | |
| r.182g>a | P3 | aplastic anemia | Vulliamy et al, 2011 | |
| r.200_201 delinsAG | NA | Pulmonary Fibrosis | Justet et al, 2021 | |
| r.202u>g | p1a | dyskeratosis congenita | Collopy et al, 2015 | |
| r.204c>g | P1a | aplastic anemia | Fogarty et al, 2003 | |
| r.205c>u | p1a | dyskeratosis congenita | Collopy et al, 2015 | |
| Hypervariable | r.212c>g | P4 | dyskeratosis congenita, myelodysplastic syndrome, and aplastic anemia | Collopy et al, 2015 Kirwan et al, 2009 |
| r.216_229del14 | P4.1 | autosomal dominant dyskeratosis congenita | Ly et al, 2005.2 | |
| r.228g>a | P4.1 | polymorphism | Yamaguchi et al, 2003 Marrone et al, 2004 |
|
| r.235C>G | Pulmonary Fibrosis | Justet et al, 2021 | ||
| r.236C>U | Pulmonary Fibrosis | Borie et al, 2016 | ||
| r.242c>u | P5 | hoyeraal hreidarsson syndrome | Vulliamy et al, 2011 | |
| CR4-CR5 | r.257g>u | P6b | bone marrow failure | Alder et al, 2018 |
| r.287c>g | P6b | myelodysplastic syndrome | Vulliamy et al, 2011 | |
| r.305g>a | P6.1 | aplastic anemia | Yamaguchi et al, 2003 | |
| r.309g>u | P6.1 | acute myeloid leukaemia | Holme et al, 2012 | |
| r.316_451del136 | J6/5 to Box ACA | autosomal dominant dyskeratosis congenita | Vulliamy et al, 2004 | |
| r.319g>a | P5 | liver fibrosis | Boyraz et al, 2016 | |
| r.322g>a | P5 | myelodysplasia and refractory anemia | Yamaguchi et al, 2003 | |
| r.323c>u | P5 | myelodysplastic syndrome | Takeughi et al, 2007 | |
| r.323C>G | Pulmonary Fibrosis | Justet et al, 2021 | ||
| r.325g>u | P5 | idiopathic pulmonary fibrosis | Alder et al, 2008 | |
| Hypervariable | r.341_360del20 | P4.1_P4.2 | pancytopenia | Calado et al, , 2009.2 |
| ScaRNA | r.375_377delgga | Box H | idiopathic pulmonary fibrosis | Alder et al, 2011 |
| r.377a>g | Box H | aplastic anemia, myelodysplastic syndrome | Vulliamy et al, 2011 Ueda et al, 2014 |
|
| r.378_451del74 | Box H to Box ACA | autosomal dominant dyskeratosis congenita | Vulliamy et al, 2001 | |
| r.389_390delcc | P7b | essential thrombocythemia | Ly et al, 2005.1 | |
| r.408c>g | P8b | autosomal dominant dyskeratosis congenita | Vulliamy et al, 2001 | |
| r.448A>U | Pulmonary Fibrosis (pneumocystosis) | Justet et al, 2021 Borie et al, 2017 |
||
| r.448A>G | Pulmonary Fibrosis | Justet et al, 2021 | ||
| r.448a>g | Box ACA | dyskeratosis congenita | Collopy et al, 2015 | |
| 3' UTR | c.467u>c | 3' UTR | aplastic anemia | Yamaguchi et al, 2003 |
TERT
TERT mutations (autosomal dominant DKC, AA, IPF)
Numerous mutations causing amino acid substitution, additions, deletions, and frame shifts within TERT, the essential protein component of the telomerase ribonucleoprotein (RNP), have been connected with human diseases. TERT contains the catalytic site for the synthesis of telomeric repeats from the RNA template. TERT is composed of three domains, N-terminal extension (NTE) that contains RNA-interaction domains 1 and 2 (RID1 and RID2), reverse transcription domain (RT) where nucleotide transfer occurs, and a C-terminal extension (CTE) for processivity and localization.
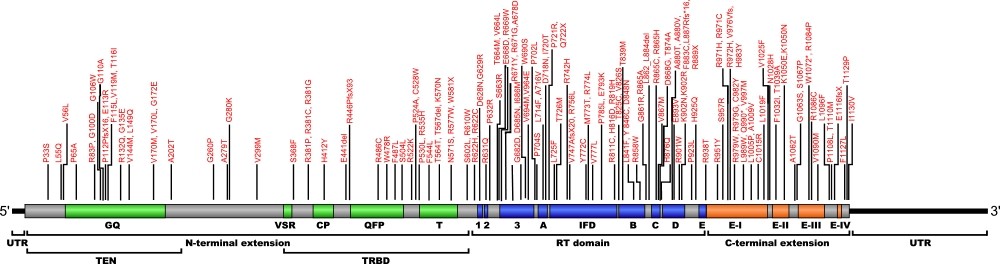
The above structural organizational scheme for the various motifs with the three domains of the TERT protein has indicated the locations of mutations known to cause human diseases. The black line represents the mRNA sequence of 4015 nt with the untranslated regions (UTR) labeled and the grey box corresponds to the protein sequence. The individual motifs are labeled and NTE is denoted by green, CTE by orange, and the central RT domain by blue boxes. The Genbank accession numbers used are NT_006576 for the 41881 bp gene and NM_198253 for the cDNA and amino acid sequences. Below is the 1132 amino acid sequence for TERT protein with the motifs labeled and colored by domain. The mutated residues are colored red and the change in amino acids is labeled.
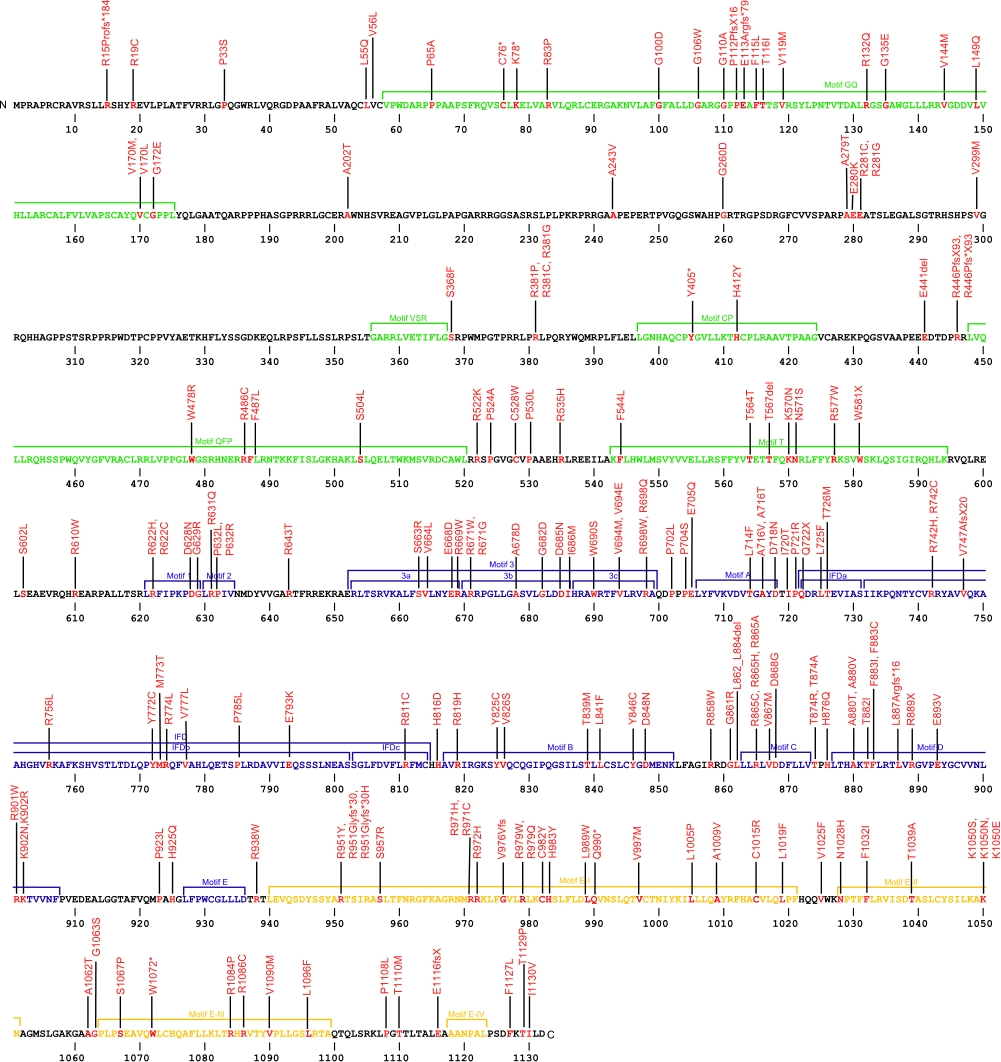
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the telomerase protein TERT. The cDNA sequence is used for the nucleotide sequence. The mutations are organized by domain from the N-terminus to the C-terminus and grouped by domain.
TERT mutations
| Domains | Mutation | AA substitution | Motif | Presentation | References |
|---|---|---|---|---|---|
| N-Terminal | c.22_43dup22 | p.Arg15Profs*184 | n/a | Pulmonary fibrosis | Borie et al, 2016 |
| c.55C>T | p.Arg19Cys | n/a | Pulmonary fibrosis | Borie et al, 2016 | |
| c.97C>T | p.Pro33Ser | n/a | idiopathic pulmonary fibrosis and fibrosis | Tsakiri et al, 2007 | |
| r.110_113delGACT | n/a | Pulmonary fibrosis | Borie et al, 2015 | ||
| c.164T>A | p.Leu55Gln | n/a | idiopathic pulmonary fibrosis | Armanios et al, 2007 | |
| c.166G>C | p.Val56Leu | n/a | aplastic anemia | Vulliamy et al, 2011 | |
| c.193C>G | p.Pro65Ala | GQ | acute myeloid leukemia | Calado et al, 2009 | |
| c.228C>A | p.Cys76* | GQ | Pulmonary fibrosis | Borie et al, 2016 | |
| c.232A>T | p.Lys78* | GQ | Pulmonary fibrosis | Justet et al, 2021 | |
| c.248G>C | p.Arg83Pro | GQ | aplastic anemia, myelodysplastic syndrome | Vulliamy et al, 2011 | |
| c.250C>T | p.(?) | GQ | head and neck ucosal melanoma | Sari et al, 2016 | |
| c.277+1G>A | n/a | IVS1 | idiopathic pulmonary fibrosis | Armanios et al, 2007 | |
| c.299G>A | p.Gly100Asp | GQ | Pulmonary fibrosis | Justet et al, 2021 | |
| c.(?) | p.Gly106Trp | GQ | cryptic dyskeratosis congenita | Yamaguchi et al, 2015 | |
| c.329G>C | p.Gly110Ala | GQ | dyskeratosis congenita | Collopy et al, 2015 | |
| c.(334_336)delC | p.Pro112ProfsX16 | GQ | idiopathic pulmonary fibrosis | Armanios et al, 2007 | |
| c. | p.Thr116Ile | GQ | Myelodysplastic syndrome | Schratz et al, 2020 | |
| c.336dupC | p.Glu113Argfs*79 | GQ | Pulmonary fibrosis | Justet et al, 2021 | |
| c.345C>G | p.Phe115Leu | GQ | Myelodysplastic syndrome and Acute myeloid leukemia | Schratz et al, 2021 | |
| c.355G>A | p.Val119Met | GQ | dyskeratosis congenita | Collopy et al, 2015 | |
| c.395G>A | p.Arg132Gln | GQ | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
|
| c.(?) | p.Gly135Glu | GQ | hepatopulmonary syndrome | Gorgy et al, 2015 | |
| c.430G>A | p.Val144Met | GQ | idiopathic pulmonary fibrosis and pleuroparenchymal fibroelastosis | Tsakiri et al, 2007 Newton et al, 2016 |
|
| c.446T>A | p.Leu149Gln | GQ | Pulmonary fibrosis | Justet et al, 2021 | |
| c.508G>A | p.Val170Met | GQ | aplastic anemia and pulmonary fibrosis | Parry et al, 2011 | |
| c.515G>A | p.Gly172Glu | GQ | Pulmonary fibrosis | Justet et al, 2021 | |
| c.(?) | p.Val170Leu | GQ | idiopathic pulmonary fibrosis | Alder et al, 2018 | |
| c.604G>A | p.Ala202Thr | n/a | aplastic anemia | Vulliamy et al, 2005 Yamaguchi et al, 2005 |
|
| c.779G>A | p.Gly260Asp | n/a | aplastic anemia | Calado et al, 2009.1 | |
| c.(?) | p.Ala243Val | n/a | hepatocellular carcinoma | Donaires et al, 2017 | |
| c.1849C>T | p.Ala279Thr | n/a | polymorphism | Du et al, 2009 Alder et al, 2008 Vulliamy et al, 2006 |
|
| c.(?) | p.Glu280Lys | n/a | dyskeratosis congenita | Yamaguchi et al, 2015 | |
| c.895G>A | p.Val299Met | n/a | acute myeloid leukemia | Calado et al, 2009 | |
| c.896G>A | p.(?) | n/a | acute myeloid leukaemia | Yan et al, 2012 | |
| c.1002_1004del | p.334_335del | n/a | dyskeratosis congenita | Yamaguchi et al, 2015 | |
| c.1062A>T | p.(?) | n/a | acute myeloid leukaemia | Aref et al, 2014 | |
| c.1079C>G | p.(?) | n/a | acute myeloid leukaemia | Yan et al, 2012 | |
| c.1103C>T | p.Ser368Phe | n/a | pancytopenia, aplastic anemia, pulmonary fibrosis | Calado et al, 2009.2 | |
| c.1141C>T | p.Arg281Cys | n/a | dyskeratosis congenita | Collopy et al, 2015 | |
| c.1141C>G | p.Arg281Gly | n/a | dyskeratosis congenita | Collopy et al, 2015 | |
| c.1142G>C | p.Arg381Pro | n/a | dyskeratosis congenita | Vulliamy et al, 2011 | |
| c.1215C>G | p.Tyr405* | CP | Pulmonary fibrosis | Justet et al, 2021 | |
| c.1234C>T | p.His412Tyr | CP | polymorphism/aplastic anemia/myelodysplastic syndrome/idiopathic pulmonary fibrosis | Alder et al, 2008 Yamaguchi et al, 2005 Juge et al, 2017 |
|
| c.1321_1323delGAG | p.Glu441del | n/a | polymorphism/hepatic cirrhosis | Yamaguchi et al, 2005 Calado et al, 2011 |
|
| c.1336_1337insC | p.Arg446ProfsX93 | n/a | dyskeratosis congenita | Collopy et al, 2015 | |
| c.1336dupC | p.Arg446Profs*93 | n/a | Dyskeratosis congenita, pulmonary fibrosis | Justet et al, 2021 | |
| c.1432T>C | p.Trp478Arg | QFP | Pulmonary fibrosis | Justet et al, 2021 | |
| c.1451G>C | p.(?) | QFP | acute myeloid leukaemia | Yan et al, 2012 | |
| c.1456C>T | p.Arg486Cys | QFP | idiopathic pulmonary fibrosis | Tsakiri et al, 2007 | |
| c.(?) | p.Phe487Leu | QFP | liver disease and bone marrow failure | Alder et al, 2018 | |
| c.1511C>T | p.Ser504Leu | QFP | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
|
| c.1565G>A | p.Arg522Lys | n/a | acute myeloid leukemia | Calado et al, 2009 | |
| c.1570C>G | p.Pro524Ala | n/a | aplastic anemia | Collopy et al, 2014 | |
| c.1584T>G | p.Cys528Trp | n/a | Pulmonary fibrosis | Snetselaar et al, 2017 Justet et al, 2021 |
|
| c.1589C>T | p.Pro530Leu | n/a | hoyeraal-hreidarsson syndrome/dyskeratosis congenita/hepatic cirrhosis/pulmonary fibrosis | Vogiatzi et al, 2013 Carrillo et al, 2012 Calado et al, 2011 Maryoung et al, 2017 |
|
| c.1604G>A | p.Arg535His | n/a | Myelodysplastic syndrome | Schratz et al, 2020 | |
| c.1630T>C | p.Phe544Leu | T | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 Borie et al, 2015 |
|
| c.1692G>A | p.Thr564Thr | T | Pulmonary fibrosis | Borie et al , 2016 | |
| c.1698_1700delCAC | p.Thr567del | T | Pulmonary fibrosis | Snetselaar et al, 2017 Justet et al, 2021 |
|
| c.1710G>C | p.Lys570Asn | T | Pulmonary fibrosis | Borie et al, 2015 Justet et al, 2021 |
|
| c.1710G>(T_C) | p.Lys570Asn | T | Heterozygote presents as aplastic anemia | Xin et al, 2007 | |
| c.1710G>(T_C) | p.Lys570Asn | T | Homozygote presents as Hoyeraal Hreidarsson syndrome | Gramatges et al , 2013 | |
| c.1729C>T | p.Arg577Trp | T | Pulmonary fibrosis | Justet et al, 2021 | |
| c.(?) | p.Asn571Ser | T | pulmonary fibrosis and bone marrow failure | Alder et al, 2018 | |
| c.1743delG | p.Trp581X | QFP | dyskeratosis congenita | Collopy et al, 2015 | |
| c.1769+1G>A | n/a | IVS | dyskeratosis congenita | Collopy et al, 2015 | |
| c.1805C>T | p.Ser602Leu | n/a | Pulmonary fibrosis | Justet et al, 2021 | |
| Reverse transcriptase | c.(?) | p.Arg622His | 1 | idiopathic pulmonary fibrosis | Dai et al, 2014 |
| c.1828C>T | p.Arg610Trp | n/a | Pulmonary fibrosis | Nunes et al, 2017 Justet et al, 2021 |
|
| c.1864C>T | p.Arg622Cys | 1 | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
|
| c.1882G>A | p.Asp628Asn | 1 | Pulmonary fibrosis | Justet et al, 2021 | |
| c.1885G>C | p.Gly629Arg | 1 | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |
| c.1892G>A | p.Arg631Gln | 2 | pulmonary fibrosis | Diaz de Leon et al, 2010 | |
| c.1895C>T | p.Pro632Leu | 2 | pulmonary fibrosis | Maryoung et al, 2017 | |
| c.1895C>G | p.Pro632Arg | 2 | cryptic dyskeratosis congenita | Yamaguchi et al, 2015 | |
| c.1928G>C | p.Arg643Thr | n/a | Myelodysplastic syndrome | Schratz et al, 2020 | |
| c.1989G>C | p.Ser663Arg | 3a | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |
| c.(?) | p.Val664Leu | 3a | bone marrow failure and idiopathic pulmonary fibrosis | Alder et al, 2018 | |
| c.2005C>T | p.Arg669Trp | 3a | Pulmonary fibrosis | Newton et al, 2016 Snetselaar et al, 2017 Van der vis et al, 2020 Justet et al, 2021 |
|
| c.2011C>T | p.Arg671Trp | 3b | Pulmonary fibrosis | Diaz de Leon et al, 2010 Snetselaar et al, 2017 Justet et al, 2021 |
|
| c. | p.Arg671Gly | 3b | Myelodysplastic syndrome | Schratz et al, 2020 | |
| c.2033C>A | p.Ala678Asp | 3b | pulmonary fibrosis | Chambers et al, 2012 | |
| c.2045G>A | p.Gly682Asp | 3b | aplastic anemia | Liang et al, 2006 | |
| c.2053G>A | p.Asp685Asn | 3b | Myelodysplastic syndrome | Schratz et al, 2020 | |
| c.2058C>G | p.Ile686Met | 3b | Myelodysplastic syndrome | Schratz et al, 2020 | |
| c.2062C>G | p.Glu668Asp | 3a | cyrptogenic cirrhosis | Valenti et al, 2013 | |
| c.2069G>C | p.Trp690Ser | 3c | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |
| c.2080G>A | p.Val694Met | 3c | Pulmonary fibrosis, Cancer | Yamaguchi et al, 2005 Schratz et al, 2020 Justet et al, 2021 |
|
| c.2081C>T | p.Val694Glu | 3c | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |
| c.2092C>T | p.Arg698Trp | 3c | dyskeratosis congenita | Carrillo et al, 2012 | |
| c.2093G>A | p.Arg698Gln | 3c | dyskeratosis congenita | Carrillo et al, 2012 | |
| c.2093G>A | p.Arg698Gln | 3c | Pulmonary fibrosis | Borie et al, 2016 | |
| c.2105C>T | p.Pro702Leu | n/a | pulmonary fibrosis | Cronkhite et al, 2008 | |
| c.2105C>T | p.Pro702Leu | n/a | Pulmonary fibrosis | Borie et al, 2016 | |
| c.2110C>T | p.Pro704Ser | n/a | autosomal dominant dyskeratosis congenita/aplastic anemia/idiopathic pulmonary fibrosis/myelodysplastic syndrome | Du et al, 2009 Du et al, 2008 Petrovski et al, 2017 Keel et al, 2016 Schratz et al, 2020 |
|
| c.2113G>C | p.Glu705Gln | n/a | dyskeratosis congenita | Collopy et al, 2015 | |
| c.2146G>A | p.Ala716Thr | A | Aplastic anemia, pulmonary fibrosis | Parry et al, 2011 Borie et al, 2016 Snetselaar et al, 2017 Justet et al, 2021 |
|
| c.2147C>T | p.Ala716Val | A | severe pancytopenia and aplastic anemia | Du et al, 2009 | |
| c.2147C>T | p.Ala716Val | A | Pulmonary fibrosis | Vulliamy et al, 2011 Justet et al, 2021 |
|
| c.2152G>A | p.Asp718Asn | A | dyskeratosis congenita | Vulliamy et al, 2011 | |
| c.2159T>C | p.Ile720Thr | n/a | Pulmonary fibrosis | Borie et al, 2016 | |
| c.2162C>G | p.Pro721Arg | n/a | autosomal recessive dyskeratosis congenita | Vulliamy et al, 2006 | |
| c.(?) | p.Gln722X | IFDa | bone marrow failure | Alder et al, 2018 | |
| c.2173C>T | p.Leu714Phe | n/a | aplastic anemia | Collopy et al, 2014 | |
| c.2177C>T | p.Thr726Met | IFDa | aplastic anemia | Liang et al, 2006 | |
| c.2224C>T, c.2655-47_2659dup | p.Arg742Cys p.Leu887Argfs*16 | n/a | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2225G>A | p.Arg742His | IFDb | Pulmonary fibrosis | Petrovski et al, 2017 Justet et al, 2021 |
|
| c.2240delT | p.Val747AlafsX20 | IFDb | idiopathic pulmonary fibrosis | Tsakiri et al, 2007 | |
| c.2267G>T | p.Arg756Leu | IFDb | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
|
| c.2287-2A>G | IFDb | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
||
| c.2287-2A>C | p.Arg756Leu | n/a | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
|
| c.2315A>G | p.Tyr772Cys | IFDb | aplastic anemia | Yamaguchi, 2005 | |
| c.2318T>C | p.Met773Thr | IFDb | dyskeratosis congenita | Collopy et al, 2015 | |
| c.2321G>T | p.Arg774Leu | IFDb | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2329G>A | p.Val777Leu | IFDb | dyskeratosis congenita, idiopathic pulmonary fibrosis | Collopy et al, 2015 Dai et al, 2014 |
|
| c.2354C>T | p.Pro785Leu | IFDb | acute myeloid leukaemia | Holme et al, 2012 | |
| c.2377G>A | p.Glu793Lys | IFDb | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2383-2A>G | n/a | IVS | idiopathic pulmonary fibrosis | Juge et al, 2017 | |
| c.2431C>T | p.Arg811Cys | IFDc | autosomal recessive dyskeratosis congenita | Marrone et al, 2007.2 | |
| c.2431C>T | p.Arg811Cys | IFDc | Pulmonary fibrosis | Marrone et al, 2007.2 Justet et al, 2021 |
|
| c.2446C>G | p.His816Asp | B | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2456G>A | p.Arg819His | B | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2468+6T>G | n/a | B | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2474A>G | p.Tyr825Cys | B | dyskeratosis congenita | Collopy et al, 2015 | |
| c.? | p.Val826Ser | B | Myelodysplastic syndrome | Schratz et al, 2020 | |
| c.2516C>T | p.Thr839Met | B | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2521C>T | p.Leu841Phe | B | pulmonary fibrosis | Parry et al, 2011 | |
| c.2537A>G | p.Tyr846Cys | B | aplastic anemia | Du et al, 2008 | |
| c.2542G>A | p.Asp848Asn | B | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2572C>T | p.Arg858Trp | n/a | pulmonary fibrosis, liver disease and bone marrow failure | Newton et al, 2016 | |
| c.2581G>A | p.Gly861Arg | n/a | myelodysplastic syndrome | Vulliamy et al, 2011 Schratz et al, 2020 |
|
| c.2583-2A>C | p.Leu862_Leu884del | IVS9 | idiopathic pulmonary fibrosis | Armanios et al, 2007 | |
| c.2593C>T | p.Arg865Cys | C | idiopathic pulmonary fibrosis | Tsakiri et al, 2007 | |
| c.2594G>A | p.Arg865His | C | idiopathic pulmonary fibrosis, fibrosis, and aplastic anemia | Tsakiri et al, 2007 Diaz de Leon et al, 2010 Newton et al, 2016 Justet et al, 2021 |
|
| c.(?) | p.Arg865Ala | C | idiopathic pulmonary fibrosis | Newton et al, 2016 | |
| c.2599G>A | p.Val867Met | C | pulmonary fibrosis | Diaz de Leon et al, 2010 | |
| c.2603A>G | p.Asp868Gly | C | dyskeratosis congenita | Sharma et al, 2014 | |
| c.(?) | p.Thr874Arg | C | idiopathic pulmonary fibrosis | Newton et al, 2016 | |
| c.2620A>G | p.Thr874Ala | C | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |
| c.2628C>G | p.His876Gln | C | aplastic anemia | Du et al, 2008 | |
| c.2638G>A | p.Ala880Thr | D | hoyeraal-hreidarsson syndrome | Vogiatzi et al, 2013 | |
| c.2638G>A | p.Ala880Thr | D | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
|
| c.2639C>T | p.Ala880Val | D | Pulmonary fibrosis | Borie et al, 2016 | |
| c.2648T>G | p.Phe883Cys | D | idiopathic pulmonary fibrosis | Fernandez et al, 2012 | |
| c.2665C>T | p.Arg889X | D | aplastic anemia | Calado et al, 2009.1 | |
| c.2672T>A | p.Phe883Ile | D | pulmonary fibrosis | Maryoung et al, 2017 | |
| c.2678A>T | p.Glu893Val | D | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
|
| c.2701C>T | p.Arg901Trp | D | autosomal recessive dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Marrone et al, 2007.2 | |
| c.2706G>C | p.Lys902Asn | D | autosomal dominant dyskeratosis congenita and aplastic anemia | Armanios et al, 2005 | |
| c.2705A>G | p.Lys902Arg | D | aplastic anemia | Parry et al, 2011 | |
| c.2747G>A | p.Arg979Gln | E-I | Myelodysplastic syndrome | Schratz et al, 2020 | |
| c:2768C>T | p.Pro923Leu | n/a | pulmonary fibrosis | Gansner et al, 2012 | |
| c.2775C>A | p.His925Gln | n/a | pulmonary fibrosis | Diaz de Leon et al, 2010 | |
| C-terminal | c.2812C>T | p.Arg938Trp | n/a | idiopathic pulmonary fibrosis | Petrovski et al, 2017 |
| c.2843+1G>A | E-I | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
||
| c.2849delC | p.Arg951Glyfs*30 | E-I | Pulmonary fibrosis | Borie et al, 2016 | |
| c.2851delC | p.Arg951Glyfs*30His | E-I | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2851C>T | p.Arg951Trp | E-I | pulmonary fibrosis/pleuroparenchymal fibroelastosis | Diaz de Leon et al, 2010 Newton et al, 2016 Justet et al, 2021 |
|
| c.2869A>C | p.Ser957Arg | E-I | pulmonary fibrosis | Cronkhite et al, 2008 | |
| c.2911C>T | p.Arg971Cys | E-I | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 Borie et al, 2015 |
|
| c.2912C>T | p.Arg971His | E-I | dyskeratosis congenita | Carrillo et al, 2012 | |
| c.2915G>A | p.Arg972His | E-I | dyskeratosis congenita | Vulliamy et al, 2011 | |
| c.2935C>T | p.Arg979Trp | E-I | aplastic anemia and dyskeratosis congenita, pulmonary fibrosis | Vulliamy et al, 2005 Xin et al, 2007 Borie et al, 2015 Borie et al, 2016 Justet et al, 2021 |
|
| c.2936G>A | p.Arg979Gln | E-I | dyskeratosis congenita | Collopy et al, 2015 | |
| c.2945G>A | p.Cys982Tyr | E-I | Pulmonary fibrosis | Borie et al, 2016 | |
| c.2966T>G | p.Leu989Trp | E-I | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2968C>T | p.Gln990* | E-I | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
|
| c.2989G>A | p.Val997Met | E-I | Pulmonary fibrosis | Justet et al, 2021 | |
| c.2991delG | p.Val976Valfs | E-I | pulmonary fibrosis | Maryoung et al, 2017 | |
| c.(?) | p.His983Tyr | E-I | Hepatopulmonary syndrome | Gorgy et al, 2015 | |
| c.3007A>G | p.Arg671Trp | E-I | Pulmonary fibrosis | Justet et al, 2021 | |
| c.3014T>C | p.Leu1005Pro | E-I | Pulmonary fibrosis | Justet et al, 2021 | |
| c.3026C>T | p.Ala1009Val | E-I | Pulmonary fibrosis, Myelodysplastic syndrome | Borie et al, 2016 Schratz et al, 2020 |
|
| c.3043C>T | p.Cys1015Arg | E-I | aplastic anemia | Du et al, 2009 | |
| c.3055C>T | p.Leu1019Phe | E-I | pulmonary fibrosis/chronic hypersensitivity pneumonitis | Cronkhite et al, 2008 Newton et al, 2016 |
|
| c.3073G>T | p.Val1025Phe | n/a | aplastic anemia | Parry et al, 2011 | |
| c.3082A>C | p.Asn1028His | E-II | dyskeratosis congenita | Vulliamy et al, 2011 | |
| c.(?) | p.Phe1032Ile | E-II | idiopathic pulmonary fibrosis | Dai et al, 2014 | |
| c.(?) | p.Thr1039Ala | E-II | bone marrow failure and liver disease | Alder et al, 2018 | |
| c.3148A>G | p.Lys1050Glu | E-III | Pulmonary fibrosis | Cronkhite et al, 2008 Diaz de Leon et al, 2010 Justet et al, 2021 |
|
| c.3150G>C | p.Lys1050Asn | E-II | dyskeratosis congenita | Collopy et al, 2015 | |
| c.3184G>A | p.Ala1062Thr | n/a | polymorphism/ acute myeloid leukemia/ aplastic anemia | Du et al, 2009 Alder et al, 2008 Calado et al, 2009 Ziegler et al, 2012 |
|
| c.3187G>A | p.Gly1063Ser | n/a | pulmonary fibrosis | Diaz de Leon et al, 2010 | |
| c.3199T>C | p.Ser1067Pro | E-III | Pulmonary fibrosis | Justet et al, 2021 | |
| c.3216G>A | p.Trp1072* | E-III | Pulmonary fibrosis | Borie et al, 2016 Justet et al, 2021 |
|
| c.3251 G>C | Arg1084Pro | E-III | pulmonary fibrosis | El-Chemaly et al, 2011 | |
| c.3256C>T | p.Arg1086Cys | E-III | connective tissue disease with usual interstital pneumonia associated | Petrovski et al, 2017 | |
| c.3268G>A | p.Val1090Met | E-III | aplastic anemia | Yamaguchi, 2005 | |
| c.3286C>T | p.Leu1096Phe | E-III | Pulmonary fibrosis | Justet et al, 2021 | |
| c.3323C>T | p.Pro1108Leu | n/a | idiopathic pulmonary fibrosis | Juge et al, 2017 | |
| c.3329C>T | p.Thr1110Met | n/a | idiopathic pulmonary fibrosis | Armanios et al, 2007 | |
| c.3346_3522del177 | p.Glu1116fsX | n/a | idiopathic pulmonary fibrosis, fibrosis, and aplastic anemia | Tsakiri et al, 2007 | |
| c.3379T>C | p.Phe1127Leu | n/a | resembles Hoyeraal Hreidarsson syndrome and autosomal recessive dyskeratosis congenita | Vulliamy et al, 2005 Vulliamy et al, 2006 |
|
| c.3385A>C | p.Thr1129Pro | n/a | dyskeratosis congenita | Stockklausner et al, 2015 | |
| c.3388A>G | p.Ile1130Val | n/a | aplastic anemia | Vulliamy et al, 2011 |
DKC1
DKC1 (dyskerin) mutations (X-linked recessive DKC)
Numerous mutations causing amino acid substitutions, deletions, and the loss of the entire exon 15 within dyskerin, a telomerase-associated protein, have been connected with human diseases. Dyskerin associates with the ScaRNA domain of the RNA component of telomerase along with Nop10, Gar1, and NHP2 proteins. In common with RNA pseudouridine synthases, dyskerin contains a TruB domain that participates in eukaryotic ribosomal RNA processing. The TruB domain is composed of two motifs, TruB I and TruB II. In addition to this are two nuclear localization (NL) signals, N-terminal and C-terminal, and the PUA, Pseudouridine synthase and Archaeosine transglycosylase, domain involved in RNA modification.
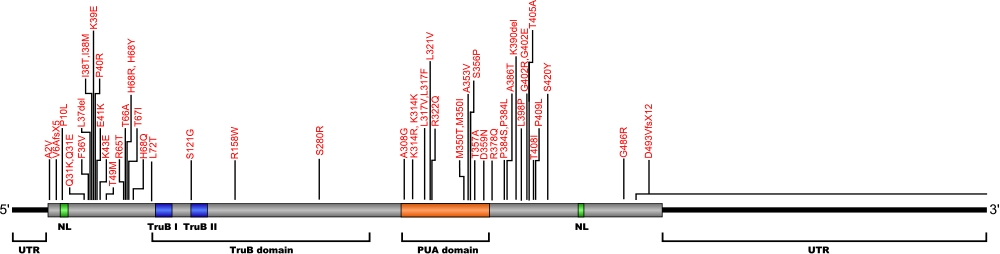
The above structural organizational scheme for the two domains, TruB and PUA, of the dyskerin protein has indicated the locations of mutations known to cause human diseases. The black line represents the mRNA sequence of 2454 nt with the untranslated regions (UTR) labeled and the grey box corresponds to the protein sequence. The N-terminal and C-terminal nuclear localization (NL) signals are denoted by green, the TruB motifs within the TruB domain by blue, and the PUA domain by orange boxes. The Genbank accession numbers used are NT_011726 for the 14811 bp gene and NM_001363 for the cDNA and amino acid sequences. Below is the 514 amino aid sequence for dyskerin protein with the NL signals, TruB motifs, and PUA domain colored and labeled. The mutated residues are colored red and the change in amino acids is labeled.
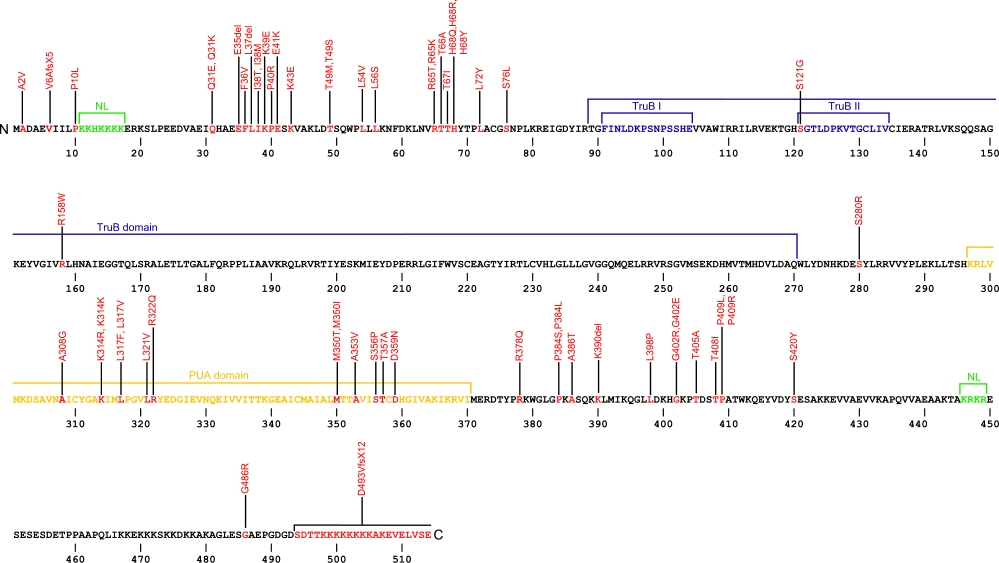
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the telomerase-associated protein dyskerin. The cDNA sequence is used for the nucleotide sequence. The mutations are organized by domain from the N-terminus to the C-terminus.
DKC1 mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| UTR | c.-141C>G | n/a | 5' UTR | X-linked recessive dyskeratosis congenita | Knight et al, 2001 |
| c.-142C>G | n/a | 5' UTR | X-linked recessive dyskeratosis congenita | Dokal et al, 2000 | |
| c.5C>T | p.Ala2Val | 1 | X-linked recessive dyskeratosis congenita | Knight et al,1999.1 | |
| c.16+592C>G r.16_17ins247, 16+343_589 | p.Val6AlafsX5 | IVS1 | X-linked recessive dyskeratosis congenita | Knight et al, 2001 | |
| c.29C>T | p.Pro10Leu | 2 | dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Vulliamy et al, 2006 | |
| c.85-5C>T | n/a | IVS2 | bone marrow failure | Alder et al, 2018 | |
| c.85-5C>G | n/a | IVS2 | X-linked recessive dyskeratosis congenita | Knight et al,1999.1 | |
| c.91C>A | p.Gln31Lys | 3 | X-linked recessive dyskeratosis congenita | Kanegane et al, 2005 | |
| c.91C>G | p.Gln31Glu | 3 | X-linked recessive dyskeratosis congenita | Wong et al, 2004 | |
| c.(?) | p.Glu35del | 3 | X-linked recessive dyskeratosis congenita | Xu et al, 2016 | |
| c.106T>G | p.Phe36Val | 3 | X-linked recessive dyskeratosis congenita | Heiss et al, 1998 | |
| c.109_111delCTT | p.Leu37del | 3 | X-linked recessive dyskeratosis congenita | Heiss et al, 1998 | |
| c.113T>C | p.Ile38Thr | 3 | X-linked recessive dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Cossu et al, 2002 | |
| c.114C>G | p.Ile38Met | 3 | dyskeratosis congenita | Vulliamy et al, 2011 | |
| c.115A>G | p.Lys39Glu | 3 | X-linked recessive dyskeratosis congenita | Knight et al,1999.1 | |
| c.119C>G | p.Pro40Arg | 3 | X-linked recessive dyskeratosis congenita | Heiss et al, 1998 | |
| c.121G>A | p.Glu41Lys | 3 | X-linked recessive dyskeratosis congenita | Knight et al,1999.1 | |
| c.127A>G | p.Lys43Glu | 3 | X-linked recessive dyskeratosis congenita | Heiss et al, 2001 | |
| c.145A>T | p.Thr49Ser | 3 | dyskeratosis congenita | Alder et al, 2013 | |
| c.146C>T | p.Thr49Met | 3 | X-linked recessive dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Knight et al,1999.2 | |
| c.(?) | p.Leu54Val | 3 | X-linked recessive dyskeratosis congenita | Xu et al, 2016 | |
| c.166_167invCT | p.Leu56Ser | 3 | X-linked recessive dyskeratosis congenita | Kurnikova et al, 2009 | |
| c.194G>A | p.Arg65Lys | 4 | X-linked recessive dyskeratosis congenita | Hisata et al, 2013 | |
| c.194G>C | p.Arg65Thr | 4 | X-linked recessive dyskeratosis congenita | Knight et al,1999.1 | |
| c.196A>G | p.Thr66Ala | 4 | X-linked recessive dyskeratosis congenita, Hoyeraal Hreidarsson syndrome and aplastic anemia | Knight et al,1999.1 Hassock et al, 1999 Keel et al, 2016 |
|
| c.200C>T | p.Thr67Ile | 4 | X-linked recessive dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Vulliamy et al, 2006 | |
| c.230A>G | p.His68Arg | 4 | X-linked recessive dyskeratosis congenita | Carrillo et al, 2012 | |
| c.202C>T | p.His68Tyr | 4 | hoyeraal hreidarrson syndrome | Vulliamy et al, 2011 | |
| c.204C>A | p.His68Gln | 4 | dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Vulliamy et al, 2006 | |
| c.214_215CT>TA | p.Leu72Tyr | 4 | X-linked recessive dyskeratosis congenita | Heiss et al, 1998 | |
| c.227C>T | p.Ser76Leu | 4 | dyskeratosis congenita | Vulliamy et al, 2011 | |
| TruB | c.361A>G | p.Ser121Gly | 5 | X-linked recessive dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Knight et al,1999.2 |
| c.472C>T | p.Arg158Trp | 6 | X-linked recessive dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Knight et al, 2001 | |
| c.838A>C | p.Ser280Arg | 9 | X-linked recessive dyskeratosis congenita | Knight et al, 2001 | |
| PUA | c.911G>A | p.Ser304Asn | 10 | X-linked recessive dyskeratosis congenita, Hoyeraal Hreidarrson syndrome | Du et al, 2009 |
| c.915+10G>A | 10 | Myelodysplastic syndrome / Acute myeloid leukemia | Schratz et al, 2021 | ||
| c.(?) | p.Ala308Gly | 10 | bone marrow fialure and primary immunodeficiency | Alder et al, 2018 | |
| c.941A>G | p.Lys314Arg | 10 | X-linked recessive dyskeratosis congenita and Hoyeraal Hreidarsson syndrome, dyskeratosis congenita | Vulliamy et al, 2006 | |
| c.942G>A | p.Lys314Lys | 10 | Idiopathic pulmonary fibrosis | Gaysinskaya et al, 2020 | |
| c.949C>G | p.Leu317Val | 10 | X-linked recessive dyskeratosis congenita | Du et al, 2009 | |
| c.949C>T | p.Leu317Phe | 10 | X-linked recessive dyskeratosis congenita | Marrone et al, 2003 | |
| c.961C>G | p.Leu321Val | 10 | X-linked recessive dyskeratosis congenita | Knight et al,1999.1 | |
| c.965G>A | p.Arg322Gln | 10 | X-linked recessive dyskeratosis congenita | Marrone et al, 2003 | |
| c.1049T>C | p.Met350Thr | 11 | X-linked recessive dyskeratosis congenita | Knight et al,1999.1 | |
| c.1050G>A | p.Met350Ile | 11 | X-linked recessive dyskeratosis congenita | Knight et al,1999.1 | |
| c.1058C>T | p.Ala353Val | 11 | X-linked recessive dyskeratosis congenita, Hoyeraal Hreidarsson syndrome, Myelodysplastic syndrome, Acute myeloid leukemia | Knight et al,1999.1 Schratz et al, 2021 |
|
| c.1069A>G | p.Thr357Ala | 11 | X-linked recessive dyskeratosis congenita | Kanegane et al, 2005 | |
| c.1066T>C | p.Ser356Pro | 11 | X-linked recessive dyskeratosis congenita | Coelho et al, 2011 | |
| c.1075G>A | p.Asp359Asn | 11 | dyskeratosis congenita | Vulliamy et al, 2006 | |
| c.1133G>A | p.Arg378Gln | 11 | Hoyeraal Hreidarrson syndrome | Vulliamy et al, 2011 | |
| c.1150C>T | p.Pro384Ser | 11 | X-linked recessive dyskeratosis congenita | Marrone et al, 2003 | |
| c.1151C>T | p.Pro384Leu | 11 | X-linked recessive dyskeratosis congenita | Knight et al, 2001 | |
| c.1156G>A | p.Ala386Thr | 12 | dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Vulliamy et al, 2006 | |
| c.1168_1179delAAG | p.Lys390del | 12 | X-linked recessive dyskeratosis congenita | Carrillo et al, 2012 | |
| c.1193T>C | p.Leu398Pro | 12 | X-linked recessive dyskeratosis congenita | Hiramatsu et al, 2002 | |
| c.1204G>A | p.Gly402Arg | 12 | X-linked recessive dyskeratosis congenita | Knight et al,1999.1 | |
| c.1205G>A | p.Gly402Glu | 12 | X-linked recessive dyskeratosis congenita | Heiss et al, 1998 | |
| c.1213A>G | p.Thr405Ala | 12 | familial interstitial pneumonia | Kropski et al, 2014 | |
| c.1223C>T | p.Thr408Ile | 12 | X-linked recessive dyskeratosis congenita | Vulliamy et al, 2006 | |
| c.1226C>T | p.Pro409Leu | 12 | X-linked recessive dyskeratosis congenita | Ding et al, 2004 | |
| c.1226C>G | p.Pro409Arg | 12 | dyskeratosis congenita | Alder et al, 2013 | |
| c.1258_1259AG>TA | p.Ser420Tyr | 12 | dyskeratosis congenita | Vulliamy et al, 2006 | |
| c.1456G>A | p.Gly486Arg | 13 | hoyeraal-hreidarsson syndrome | Vogiatzi et al, 2013 | |
| c.1477-2A>G | n/a | IVS14 | dyskeratosis congenita | Vulliamy et al, 2006 | |
| c.1476+51_oMPP1:c.(?)del | p.Asp493ValfsX12 | 15 | X-linked recessive dyskeratosis congenita | Vulliamy et al, 1999 |
Nola2
Nola2 (NHP2) mutations (autosomal recessive DKC)
Three point mutations have been discovered within Nola2, the gene that encodes for the protein NHP2, a member of the H/ACA snoRNPs that are involved in various aspects of rRNA processing and modification. NHP2 associates with the ScaRNA domain of the RNA component of telomerase along with dyskerin, Gar1, and Nop10 proteins. These are the first mutations found within NHP2 that is known to cause human disease. The mutation c. 460T>A causes X154R +51aa, the replacement of the stop codon with arginine and the predicted addition of 51 amino acids.

The above structural organizational scheme for the four exons that form the 4498 bp Nola2 gene has indicated the location of mutations known to cause human disease. The black line represents the genomic DNA sequence with the untranslated regions (UTR) labeled. Colored boxes indicate exons 1 though 4. The Genbank accession numbers used are NT_023133 for the genomic DNA sequence and NM_017838 for the cDNA and amino acid sequences. Below is the 153 amino acid sequence for Nhp2 protein with the four exons colored and labeled. The mutated residues are colored red and the change in amino acid is labeled.
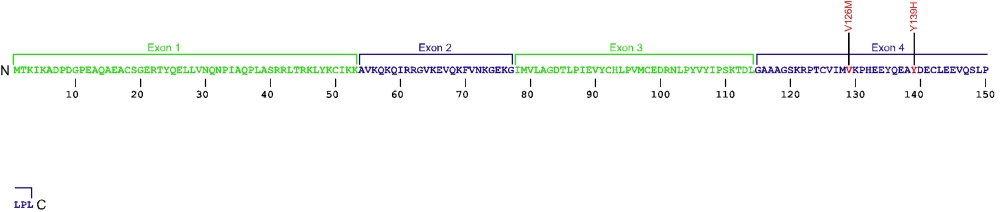
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutation within the telomerase-associated protein NHP2. The cDNA sequence is used for the nucleotide sequence.
Nola2 (NHP2) mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| c.376G>A | p.Val126Met | 4 | autosomal recessive dyskeratosis congenita | Vulliamy et al, 2008 | |
| c.415T>C | p.Tyr139His | 4 | autosomal recessive dyskeratosis congenita | Vulliamy et al, 2008 | |
| c.122G>A | p.Arg41His | 1 | BMF and PF | Benyelles et al, 2020 | |
| c.182G>C | p.Arg61Pro | 2 | HH (P1) | Benyelles et al, 2020 | |
| c.259C>T | p.Pro87Ser | 3 | HH (P1) | Benyelles et al, 2020 | |
| c.289_290del | p.Met97Valfs*2 | 3 | PF | Benyelles et al, 2020 | |
| 3' UTR | c.460T>A | p.X154Argins51 | 3' UTR | autosomal recessive dyskeratosis congenita | Vulliamy et al, 2008 |
Nola3
Nola3 (Nop10) mutations (autosomal recessive DKC)
At present only a single amino acid substitution mutation has been discovered within Nola3, the gene that encodes for the protein Nop10, a member of the H/ACA snoRNPs that are involved in various aspects of rRNA processing and modification. Nop10 associates with the ScaRNA domain of the RNA component of telomerase along with dyskerin, Gar1, and NHP2 proteins. This is the first and only mutation found within Nop10 that is known to cause human disease.

The above structural organizational scheme for the two exons that form the 1446 bp Nola3 gene has indicated the location of the mutation known to cause human disease. The black line represents the genomic DNA sequence with the untranslated regions (UTR) labeled. Colored boxes indicate exons 1 and 2. The Genbank accession numbers used are NT_010194 for the genomic DNA sequence and NM_018648 for the cDNA and amino acid sequences. Below is the 64 amino acid sequence for Nop10 protein with the two exons colored and labeled. The mutated residue is colored red and the change in amino acid is labeled.

Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutation within the telomerase-associated protein Nop10. The cDNA sequence is used for the nucleotide sequence.
Nola3 (Nop10) mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| c.100C>T | p.Arg34Trp | 2 | autosomal recessive dyskeratosis congenita | Walne et al, 2007 | |
| c.17A>G | p.Tyr6Cys | 2 | PF | Kannengiesser et al, 2020 |
WRAP53
WRAP53 (TCAB1) mutations (autosomal recessive DKC)
Currently, four point mutations causing amino acid substitutions have been documented within WRD79, the gene encoding for telomerase Cajal body protein 1 (TCAB1), a protein essential for telomerase localization to Cajal bodies and the biogenesis of the ribonucleoprotein complex. TCAB1 binds to the CAB box motif located within conserved region 7 (CR7) on the telomerase RNA (TR). Loss of function of TCAB1 results in retention of the TR at the nucleoli, inhibiting telomere extension. The mutations within TCAB1 are the first defects associated with telomerase trafficking known to cause human disease.
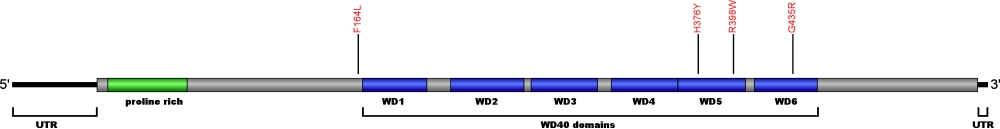
The above structural organizational scheme of telomerase Cajal body protein 1 contains a Proline rich region and six WD40 motifs (WD1-6) has indicated the locations of mutations known to cause human diseases. The black line represents the mRNA sequence of 1877 nt with the untranslated regions (UTR) labeled and the grey box corresponds to the protein sequence. The Proline rich region is denoted by green and the WD40 motifs by blue boxes. The Genbank accession numbers used are NM_001143992 for the cDNA and amino acid sequences. Below is the 548 amino aid sequence for telomerase Cajal body protein 1 with the Proline rich region and WD40 motifs colored and labeled. The mutated residues are colored red and the change in amino acids is labeled.
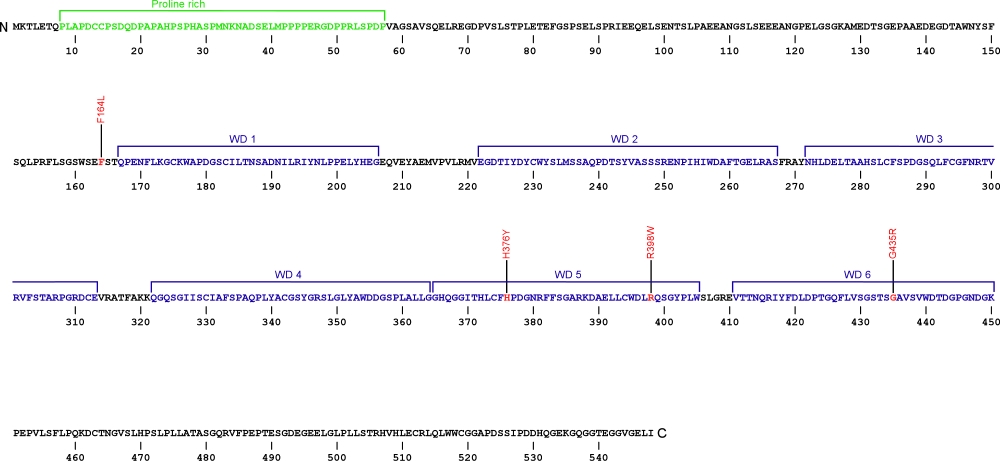
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within telomerase Cajal body protein 1. The cDNA sequence is used for the nucleotide sequence. The mutations are organized by domain from the N-terminus to the C-terminus.
WRAP53 (TCAB1) mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| c.(?) | p.Phe164Leu | 2 | autosomal recessive dyskeratosis congenita | Zhong et al, 2011 | |
| WD40 | c.1126C>T | p.His376Tyr | 7 | autosomal recessive dyskeratosis congenita | Zhong et al, 2011 |
| c.1192C>T | p.Arg398Trp | 8 | autosomal recessive dyskeratosis congenita | Zhong et al, 2011 | |
| c.(?) | p.Gly435Arg | 9 | autosomal recessive dyskeratosis congenita | Zhong et al, 2011 |
PARN
PARN mutations (familial and idiopathic pulmonary fibrosis)
Currently, 29 mutations causing amino acid substitutions, deletions and frame shifts have been documented within PARN, the gene encoding for the poly(A)-specific ribonuclease protein (PARN). The protein is a 3'-enxoribonuclease that degrades poly(A) tails of mRNAs. The mutations in the PARN protein are shown to cause the same genetic diseases as many other telomerase related proteins.
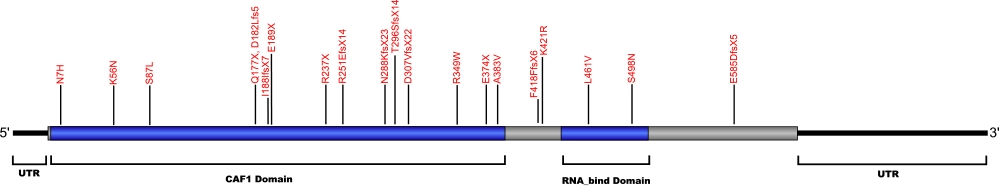
The above structural organizational scheme of poly(A)-specific ribonuclease is organized into two domains, CAF1 and the RNA_bind domain. The black line represents the mRNA sequence of 3083 nt with the untranslated regions (UTR) labeled and the grey box corresponds to the protein sequence. Both the CAF1 domain and the RNA_bind domain are labeled and indicated by solid blue boxes. The Genbank accession numbers used are NM_002582.3 for the cDNA and amino acid sequences. Below is the 639 amino aid sequence for the poly(A)-specific ribonuclease with the CAF1 and RNA_bind domains colored and labeled. The mutated residues are colored red and the change in amino acids is labeled.
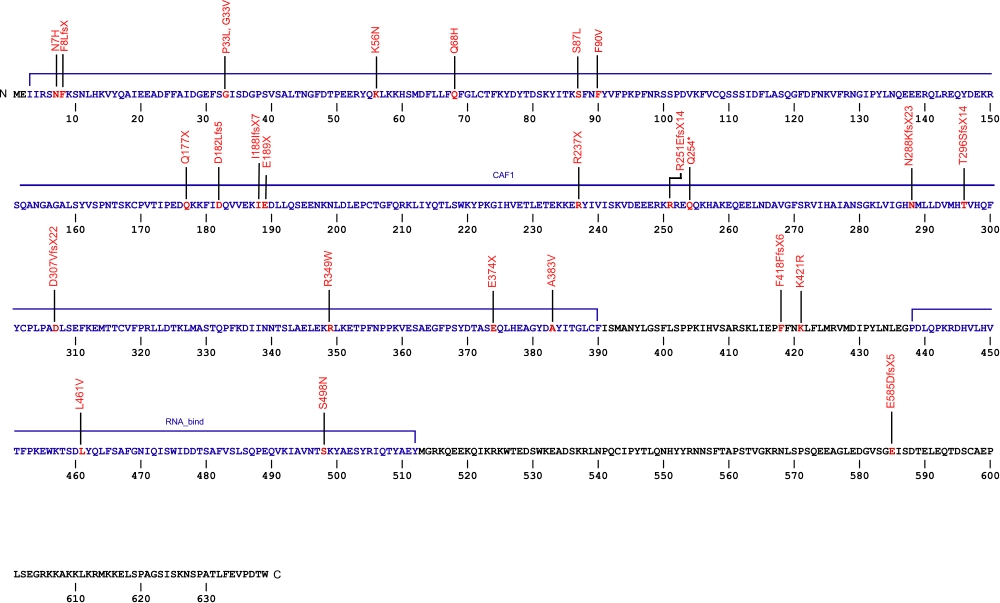
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the poly(A)-specific ribonuclease protein. The cDNA sequence is used for the nucleotide sequence. The mutations are organized by domain from the N-terminus to the C-terminus.
PARN mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| NC_000016.9:g.(?_14725823)_(14643928_?)del | Exon 1-21 del | Myelodysplastic syndrome and Acute myeloid leukemia | Schratz et al, 2021 Feurstein et al, 2020 |
||
| c.(?_-135_(c.1480+1_1481-1)del | Exon 1-21 del | Myelodysplastic syndrome and Acute myeloid leukemia | Schratz et al, 2021 | ||
| 5' UTR | c.-63C>T | n/a | 5' UTR | hoyeraal-hreidarsson syndrome | Burris et al, 2016 |
| CAF1 | heterozygous deletion Chr16:14,703,940–14,712,231 | 6 and 7 | Høyeraal-Hreidarsson (P2) | Benyelles et al, 2019 | |
| c.19A>C | p.Asn7His | dyskeratosis congenita and familial pulmonary fibrosis | Moon et al, 2014 Kropski et al, 2017 |
||
| c.(?) | p.Phe8LeufsX | idiopathic pulmonary fibrosis | Alder et al, 2018 | ||
| c.98C>T | p.Pro33Leu | Pulmonary Fibrosis | Justet et al, 2021 | ||
| c.98C>T | p.Gly33Val | Pulmonary Fibrosis | Justet et al, 2021 | ||
| c.1251delT | p.Phe418PhefsX6 | familial pulmonary fibrosis | Kropski et al, 2017 | ||
| c.168G>C | p.Lys56Asn | familial pulmonary fibrosis | Kropski et al, 2017 | ||
| c.178-3C>T | n/a | IVS | familial pulmonary fibrosis | Kropski et al, 2017 | |
| c.204G>T | p.Gln68His (homozygous) | Høyeraal-Hreidarsson (P2) | Benyelles et al, 2019 | ||
| c.245+75_245+77delCCC | n/a | IVS4 | familial pulmonary fibrosis | Kropski et al, 2017 | |
| c.246-2A>G | n/a | IVS4 | interstitial pneumonia with autoimmune features and idiopathic pulmonary fibrosis | Newton et al, 2016 Stuart et al, 2015 |
|
| c.260C>T | p.Ser87Leu | dyskeratosis congenita | Moon et al, 2014 | ||
| c.(?) | p.Phe90Val | idiopahtic pulmonary fibrosis | Alder et al, 2018 | ||
| c.529C>T | p.Gln177X | idiopathic pulmonary fibrosis and familial pulmonary fibrosis | Xing et al, 2016 Stuart et al, 2015 |
||
| c.543_544insTT | p.Asp182Leufs5 | Myelodysplastic syndrome and Acute myeloid leukemia | Schratz et al, 2021 | ||
| c.563_564insT | p.Ile188IlefsX7 | idiopathic pulmonary fibrosis | Stuart et al, 2015 | ||
| c.565G>T | p.Glu189X | familial pulmonary fibrosis | Kropski et al, 2017 | ||
| c.602+5G>A | n/a | IVS | familial pulmonary fibrosis | Kropski et al, 2017 | |
| c.659+4_659+7delAGTA | p.208_220del | IVS | dyskeratosis congenita | Tummala et al, 2015 | |
| c.703-11_703-10delAT | n/a | IVS | familial pulmonary fibrosis | Kropski et al, 2017 | |
| c.709C>T | p.Arg237X | hoyeraal-hreidarsson syndrome and non-specific interstital pneumonia | Burris et al, 2016 Petrovski et al, 2017 |
||
| c.751delA | p.Arg251GlufsX14 | idiopathi pulmonary fibrosis | Stuart et al, 2015 | ||
| c.760C>T (heterozygous) | p.Gln254* | E-I | Høyeraal-Hreidarsson (P2) | Benyelles et al, 2019 | |
| c.840+6T>C | n/a | IVS | familial pulmonary fibrosis | Kropski et al, 2017 | |
| c.863dupA | p.Asn288LysfsX23 | dyskeratosis congenita | Tummala et al, 2015 | ||
| c.887_888delCA | p.Thr296SerfsX14 | familial pulmonary fibrosis | Kropski et al, 2017 | ||
| c.918+1G>T | p.281_306del | 13 skipped | dyskeratosis congenita | Tummala et al, 2015 | |
| c.919_1262del344 | p.Asp307ValfsX22 | 14-18 | inherited bone marrow failure | Dhanraj et al, 2015 | |
| c.1006-11G>A | n/a | IVS15 | familial pulmonary fibrosis | Kropski et al, 2017 | |
| c.1045C>T | p.Arg349Trp | 16 | inherited bone marrow failure | Dhanraj et al, 2015 | |
| c.(?) g.14676110C>A | p.Glu374X | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | ||
| c.1148C>T | p.Ala383Val | dyskeratosis congenita | Tummala et al, 2015 | ||
| c.1214G>C | p.Ser405Thr | Pulmonary Fibrosis | Justet et al, 2021 | ||
| c.1262A>G | p.Lys421Arg | idiopathic pulmonary fibrosis | Stuart et al, 2015 | ||
| RNA_bind | c.(?) g.14647946G>C | p.Leu461Val | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |
| c.1493G>A | p.Ser498Asn | familial pulmonary fibrosis | Kropski et al, 2017 | ||
| c.(?) g.14540858CCT>C | p.Glu585AspfsX5 | idiopathic pulmonary fibrosis | Petrovski et al, 2017 |
TINF2
TINF2 (TIN2) mutations (autosomal dominant DKC)
Currently, three point mutations causing two amino acid substitutions have been documented within TRF1-interacting nuclear factor 2 (TINF2), the gene that encodes for the protein TIN2, a component of the shelterin complex. The shelterin complex is composed of six proteins, three of which that directly associate with the telomeric DNA, TRF1, TRF2, and POT1. Three additional proteins associate with the telomeric DNA binding proteins, TIN2, TPP1, and Rap1. The mutations within TIN2 are the first mutations found within the shelterin complex known to cause human diseases.
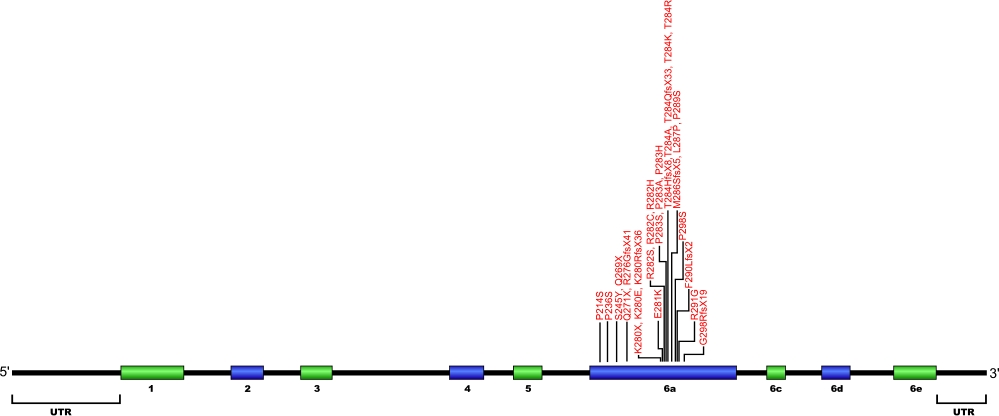
The above structural organizational scheme for the nine exons that form the 3032 bp TINF2 gene has indicated the location of mutations known to cause human diseases. The black line represents the genomic DNA sequence with the untranslated regions (UTR) labeled. Colored boxes indicate exons 1-5, 6a, 6c, 6d, and 6e. The Genbank accession numbers used are NT_026437 for the genomic DNA sequence and NM_001099274 for the cDNA and amino acid sequences. Below is the 451 amino acid sequence for TIN2 protein with the nine exons colored and labeled. The mutated residues are colored red and the change in amino acids is labeled above.
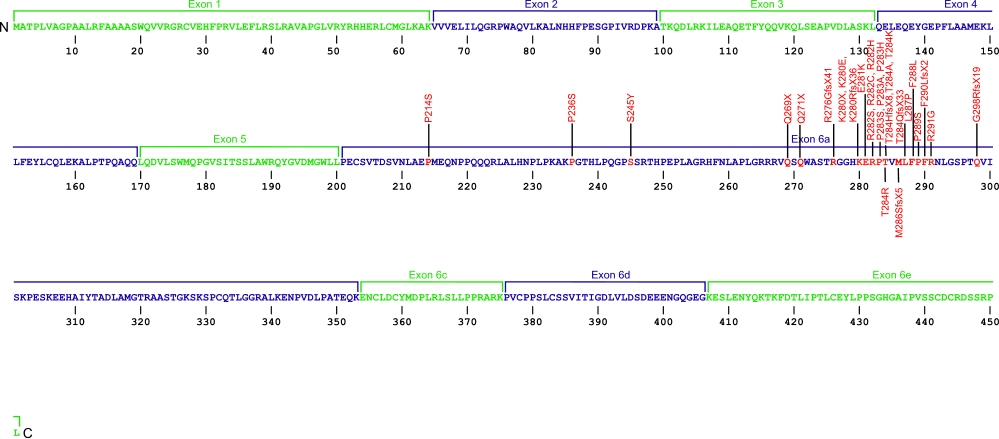
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the shelterin protein TIN2. The cDNA sequence is used for the nucleotide sequence. The mutations are organized by domain from the N-terminus to the C-terminus.
TINF2 (TIN2) mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| c.640C>T | p.Pro214Ser | 6a | bone marrow failure | Vulliamy et al, 2012 | |
| c.706C>T | p.Pro236Ser | 6a | aplastic anemia | Walne et al, 2008 | |
| c.734C>A | p.Ser245Tyr | 6a | aplastic anemia | Walne et al, 2008 | |
| c.811C>T | p.Gln271X | 6a | aplastic anemia | Vulliamy et al, 2011 | |
| c.805C>T | p.Gln269X | 6a | mucocutaneous features, bone marrow failure, dyskeratosis congenita | Vulliamy et al, 2012 Sasa et al, 2012 |
|
| c.826delA | p.Arg276GlyfsX41 | 6a | nail distrophy, bone marrow failure, lichenoid tongue, dry skin, intrauterine frowth retardation | Vulliamy et al, 2012 | |
| c.838A>T | p.Lys280X | 6a | autosomal-dominant dyskeratosis congenita, Hoyeraal Hreidarsson syndrome, and Revesz syndrome | Walne et al, 2008 | |
| c.838A>G | p.Lys280Glu | 6a | autosomal-dominant dyskeratosis congenita, Myelodysplastic syndrome and Acute myeloid leukemia | Savage et al, 2008 Schratz et al, 2020 |
|
| c.839delA | p.Lys280ArgfsX36 | 6a | dyskeratosis congenita, revesz syndrome | Sasa et al, 2012 | |
| c.841G>A | p.Glu281Lys | 6a | low wbc | Walne et al, 2008 | |
| c.844C>A | p.Arg282Ser | 6a | autosomal-dominant dyskeratosis congenita, Revesz syndrome | Savage et al, 2008 | |
| c.844C>T | p.Arg282Cys | 6a | autosomal-dominant dyskeratosis congenita and aplastic anemia | Walne et al, 2008 | |
| c.845G>A | p.Arg282His | 6a | autosomal-dominant dyskeratosis congenita, Hoyeraal Hreidarsson syndrome, and Revesz syndrome | Savage et al, 2008 | |
| c.847C>T | p.Pro283Ser | 6a | autosomal-dominant dyskeratosis congenita and Hoyeraal Hreidarsson syndrome | Walne et al, 2008 | |
| c.847C>G | p.Pro283Ala | 6a | autosomal-dominant dyskeratosis congenita | Walne et al, 2008 | |
| c.848C>A | p.Pro283His | 6a | autosomal-dominant dyskeratosis congenita | Walne et al, 2008 | |
| c.849delC | p.Thr284GlnfsX33 | 6a | nail distrophy, bone marrow failure | Vulliamy et al, 2012 | |
| c.849_850insC | p.Thr284HisfsX8 | 6a | autosomal-dominant dyskeratosis congenita and aplastic anemia | Walne et al, 2008 | |
| c.850A>G | p.Thr284Ala | 6a | autosomal-dominant dyskeratosis congenita | Walne et al, 2008 | |
| c.851C>A | p.Thr284Lys | 6a | dyskeratosis congenita | Vulliamy et al, 2012 | |
| c.851C>G | p.Thr284Arg | 6a | bone marrow failure, hair loss, dental loss, pulmonary disease, short stature, osteoporosis | Vulliamy et al, 2012 | |
| c.857delTinsGC | p.Met286SerfsX5 | 6a | nail distrophy, bone marrow failure, microcephaly, low immunoglobulins | Vulliamy et al, 2012 | |
| c.860T>C | p.Leu287Pro | 6a | autosomal-dominant dyskeratosis congenita | Walne et al, 2008 | |
| c.865_866delinsAG | p.Pro289Ser | 6a | autosomal-dominant dyskeratosis congenita | Walne et al, 2008 | |
| c.867_868insC | p.Phe290LeufsX2 | 6a | autosomal-dominant dyskeratosis congenita | Walne et al, 2008 | |
| c.871A>G | p.Arg291Gly | 6a | autosomal-dominant dyskeratosis congenita | Walne et al, 2008 | |
| c.892delC | p.Gln298ArgfsX19 | 6a | autosomal-dominant dyskeratosis congenita | Walne et al, 2008 |
TPP1
TPP1 mutations
Currently, seven amino acid substitutions and one deletion have been identified in the ACD gene. The ACD gene encodes the protein TPP1, which is a component of the shelterin complex. The shelterin complex consists of six proteins which are encoded by the genes POT1, ACD, TERF1, TERF2, TERF2IP, and TINF2. This complex helps to protect chromosome ends and is necessary for telomere functions.
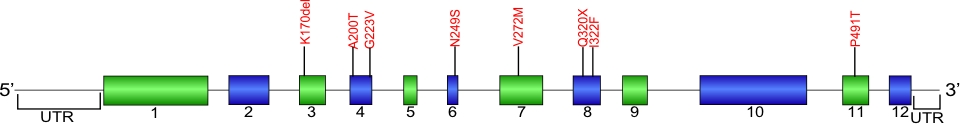
The above structural organizational scheme for the 12 exons that form the 3304 bp ACD gene has indicated the location of mutations known to cause human diseases. The black line represents the genomic DNA sequence with the untranslated regions (UTR) labeled. Colored boxes indicate exons1-12. The Genbank accession numbers used are NG_042874 for the genomic DNA sequence and NM_001082486 for the cDNA and amino acid sequence. Below is the 544 amino acid sequence for TPP1 protein with the 12 exons colored and labeled. The mutated residues are colored red and the change in amino acids is labeled above.
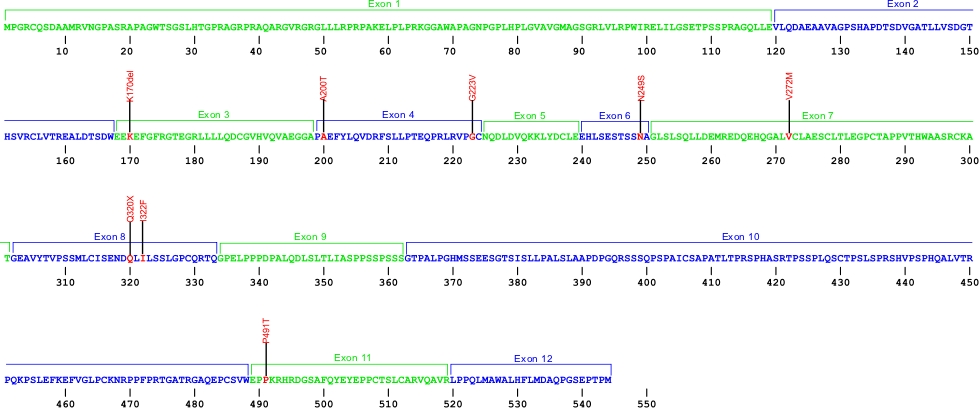
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the shelterin protein TPP1.The cDNA sequence is used for the nucleotide sequence. The mutations are organized by domain from the N-terminus to the C-terminus.
TPP1 mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| c.499_501del | p.Lys170del | 3 | Aplastic Anemia | Guo et al, 2014 | |
| c.499_501del | p.Lys170del | 3 | Hoyeraal-Hreidarsson syndrome | Kocak et al, 2014 | |
| c.598G>A | p.Ala200Thr | 4 | familial melanoma | Aoude et al, 2014 | |
| c.(?) | p.Gly223Val | 4 | lymphoblastic leukemia (somatic mutation) | Spinella et al, 2015 | |
| c.746A>G | p.Asn249Ser | 6 | familial melanoma | Aoude et al, 2014 | |
| c.814G>A | p.Val272Met | 7 | familial melanoma | Aoude et al, 2014 | |
| c.985C>T | p.Gln320X | 8 | familial melanoma | Aoude et al, 2014 | |
| c.964A>T | p.Ile322Phe | 8 | familial melanoma | Aoude et al, 2014 | |
| c.1471C>A | p.Pro491Thr | 11 | Hoyeraal-Hreidarsson syndrome | Kocak et al, 2014 |
POT1
POT1 mutations (chronic lymphocytic leukemia)
The protector of telomeres 1 (POT1) protein is a protein in the telombin family is involved in telomere maintenance and protection. The POT1 protein functions as a part of a larger protein complex that binds to TTAGGG telomere repeats in humans. The protein protects telomere DNA from recombination with other DNA, regulates telomere length and contributes to overall chromosome stability.
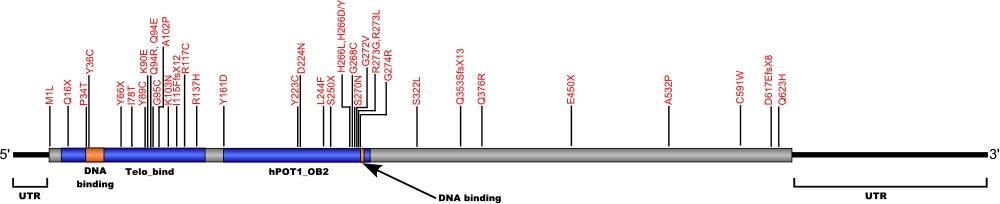
The above structural organizational scheme for the protection of telomeres 1, transcript variant 1 gene has indicated the location of mutations known to cause human diseases. The black line represents the genomic DNA sequence with the untranslated regions (UTR) labeled. Colored boxes indicate various regions identified within the POT1 protein. The Genbank accession numbers used are NM_015450.2 for the cDNA and amino acid sequences. Below is the 634 amino acid sequence for the POT1 protein with the various protein domains colored and labeled. The mutated residues are colored red and the change in amino acids is labeled above.
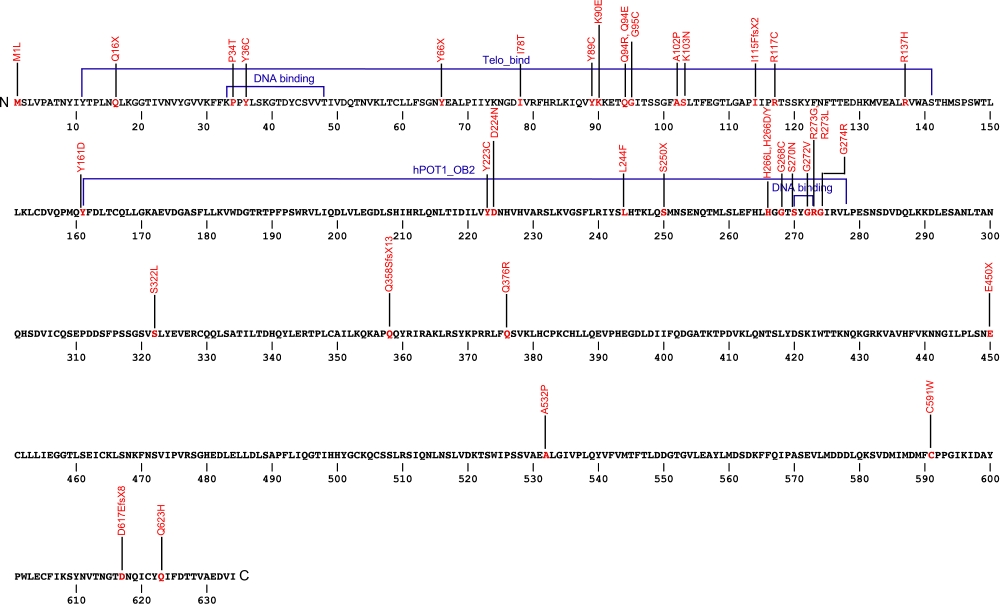
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the POT1 protein. Both the cDNA sequence and genomic position are used for the nucleotide sequence. The mutations are organized by domain from the N-terminus to the C-terminus.
POT1 mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| c.(?) g.124537227T>A | p.Met1Leu | 1 | chronic lymphocytic leukemia | Ramsay et al, 2012 | |
| Telo bind | c.(?) | p.Gln16X | 1 | chronic lymphocytic leukemia | Herling et al, 2016 |
| c.(?) | p.Pro34Thr | chronic lymphocytic leukemia | Herling et al, 2016 | ||
| c.(?) g.124532338A>T | p.Tyr36Asn | 6 | chronic lymphocytic leukemia | Ramsay et al, 2012 | |
| c.107A>G | p.Tyr36Cys | 6 | chronic lymphocytic leukemia | Speedy et al, 2016 | |
| c.(?) g.124511022A>C | p.Tyr66X | 7 | chronic lymphocytic leukemia | Ramsay et al, 2012 | |
| c.233T>C | p.Ile78Thr | familial melanoma | Wong et al, 2018 | ||
| c.266A>G | p.Tyr89Cys | familial melanoma | Robles-Espinoza et al, 2013 | ||
| c.(?) g.124503682T>C | p.Lys90Glu | 8 | chronic lymphocytic leukemia | Ramsay et al, 2012 | |
| c.268A>G | p.Lys90Glu | 8 | coats plus syndrome | Wilson et al, 2017 | |
| c.(?) g.124503669T>C | p.Gln94Arg | 8 | chronic lymphocytic leukemia | Ramsay et al, 2012 | |
| c.280C>G | p.Gln94Glu | 8 | familial melanoma | Robles-Espinoza et al, 2013 | |
| c.(?) g.124503667 | p.Gly95Cys | familial glioma | Bainbridge et al, 2015 | ||
| c.(?) | p.Ala102Pro | chronic lymphocytic leukemia | Herling et al, 2016 | ||
| c.(?) g.124499011C>A | p.Lys103Asn | lymphocytic leukemia | Kim et al, 2016 | ||
| c.(?) | p.Ile115PhefxX2 | chronic lymphocytic leukemia | Herling et al, 2016 | ||
| c.(?) g.124503601G>A | p.Arg117Cys | cardiac angiosarcoma | Calvete et al, 2015 | ||
| c.(?) g.124503540C>T | p.Arg137His | familial cutaneous malignant melanoma | Shi et al, 2013 | ||
| OB2 | c.(?) | p.Tyr161Asp | chronic lymphocytic leukemia | Herling et al, 2016 | |
| c.(?) g.124499045T>C | p.Tyr223Cys | 9 | chrnoic lymphocytic leukemia | Ramsay et al, 2012 | |
| c.(?) g.124499043C>T | p.Asp224Asn | familial cutaneous malignant melanoma | Shi et al, 2013 | ||
| c.(?) | p.Leu244Phe | chronic lymphocytic leukemia | Herling et al, 2016 | ||
| c.(?) g.124493146G>T | p.Ser250X | 10 | chronic lymphocytic leukemia | Ramsay et al, 2012 | |
| c.(?) g.124493098t>A | p.His266Leu | 10 | chronic lymphocytic leukemia | Ramsay et al, 2012 | |
| c.(?) | p.His266Asp/Tyr | 10 | chronic lymphocytic leukemia | Herling et al, 2016 | |
| c.(?) | p.Gly268Cys | 10 | chronic lymphocytic leukemia | Herling et al, 2016 | |
| c.(?) g.124493086C>T | p.Ser270Asn | 10 | familial cutaneous malignant melanoma | Shi et al, 2013 | |
| c.(?) g.124493080C>A | p.Gly272Val | 10 | chronic lymphocytic leukemia | Ramsay et al, 2012 | |
| c.(?) | p.Arg273Gln | 10 | chronic lymphocytic leukemia | Herling et al, 2016 | |
| c.818G>T | p.Arg273Leu | 10 | familial melanoma | Robles-Espinoza et al, 2013 | |
| c.(?) | p.Gly274Arg | chronic lymphocytic leukemia | Herling et al, 2016 | ||
| c.965C>T | p.Ser322Leu | coats plus syndrome | Takai et al, 2015 | ||
| c.1071_1072insT | p.Gln358SerfsX13 | chronic lymphocytic leukemia | Speedy et al, 2016 | ||
| c.1127A>G | p.Gln376Arg | chronic lymphocytic leukemia | Speedy et al, 2016 | ||
| c.1164-1G>A | n/a | IVS | chronic lymphocytic leukemia | Speedy et al, 2016 | |
| c.(?) g.124481048C>A | p.Glu450X | familial glioma | Bainbridge et al, 2015 | ||
| c.(?) g.124469308C>G | p.Ala532Pro | familial cutaneous malignant melanoma | Shi et al, 2013 | ||
| c.(?) g.12446532A>C | p.Cys591Trp | 18 | chronic lymphocytic leukemia | Ramsay et al, 2012 | |
| c.(?) g.124464068TTA>T | p.Asp617GlufsX8 | familial glioma | Bainbridge et al, 2015 | ||
| c.(?) g.124464052C>G | p.Gln623His | familial cutaneous malignant melanoma | Shi et al, 2013 |
CTC1
CTC1 mutations
Numerous mutations causing amino acid substitions, deletions, and frame shifts have been found within CTC1,a component of the CST complex. The CST complex, which is composed of the proteins CTC1, STN1, and TEN1, is involved in maintaining telomeres as well as recruiting and activating DNA polα-primase. These are the first mutations found within CTC1 that is known to cause human diseases.
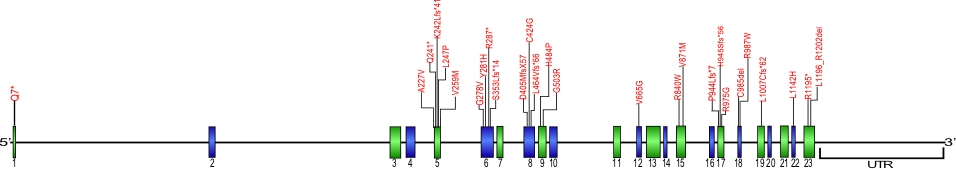
The above structural organizational scheme for the nine exons that form the 23,275 bp CTC1 gene has indicated the location of mutations known to cause human diseases. The black line represents the genomic DNA sequence with the untranslated regions (UTR) labeled. Colored boxes indicate exons 1-23.The Genbank accession numbers used are NG_032148.2 for the genomic DNA sequence and NM_025099.5 for the cDNA and amino acid sequences. Below is the 1217 amino acid sequence for CTC1 protein with 23 exons colored and labeled. The mutated residues are colored red and the change in amino acids is labeled above.
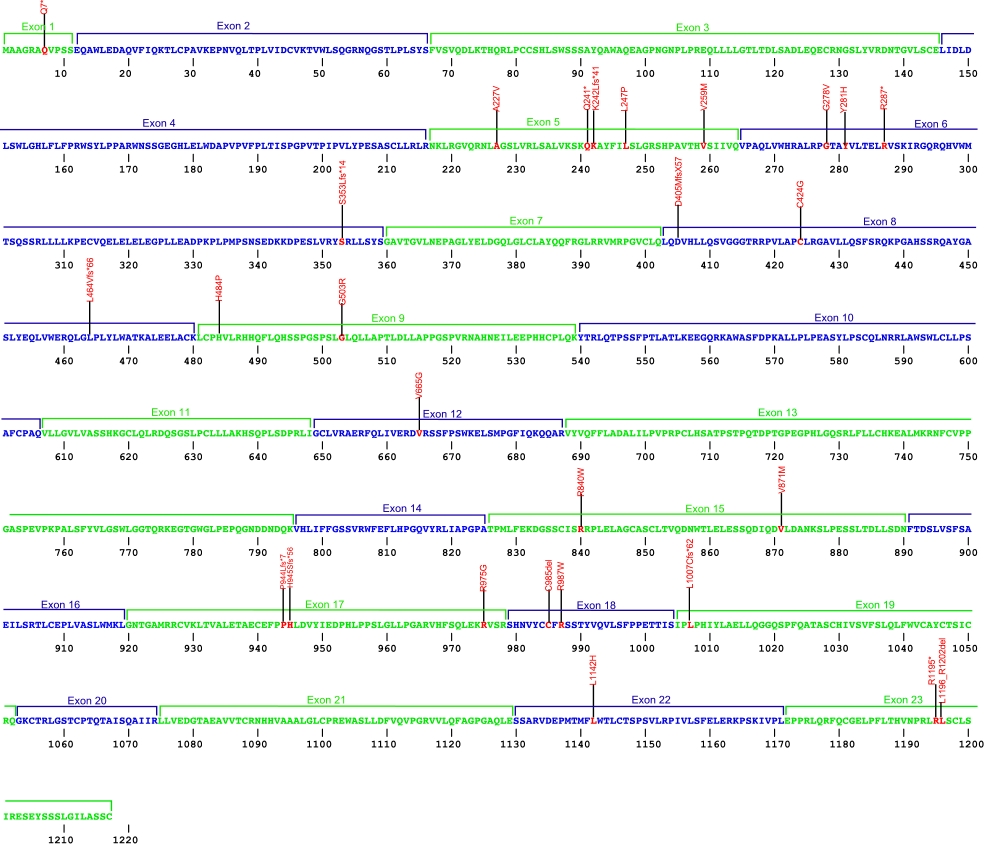
Below is a description of the locations within the nucleotide adn protein sequence, the amino acid substitions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the protein CTC1. The cDNA sequence is used for the nucleotide sequence. The mutations are organized by the domain frmo the N-terminus to the C-terminus.
CTC1 mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| c.19C>T | p.Gln7* | 1 | Coats plus | Anderson et al, 2012 | |
| c.680C>T | p.Ala227Val | 5 | cerebroretinal microangiopathy | Polvi, et al, 2012 | |
| c.721C>T | p.Gln241* | 5 | Coats plus | Anderson et al, 2012 | |
| c.721C>T | p.Gln241* | 5 | dyskeratosis congenita | Walne et al, 2013 | |
| c.724_727delAAAG | p.Lys242Leufs*41 | 5 | Coats plus | Anderson et al, 2012 | |
| c.724_727delAAAG | p.Lys242Leufs*41 | 5 | cerebroretinal microangiopathy | Polvi et al, 2012 | |
| c.724_727delAAAG | p.Lys242Leufs*41 | 5 | Dyskeratosis Congenita | Keller et al, 2012 | |
| c.724_727delAAAG | p.Lys242Leufs*41 | 5 | dyskeratosis congenita | Walne et al, 2013 | |
| c.740T>C | p.Leu247Pro | 5 | dyskeratosis congenita | Walne et al, 2013 | |
| c.775G>A | p.Val259Met | 5 | Coats plus | Anderson et al, 2012 | |
| c.775G>A | p.Val259Met | 5 | Norrie disease, cerebroretinal microangiopathy | Romaniello et al, 2012 | |
| c.833G>T | p.Gly278Val | 6 | dyskeratosis congenita | Walne et al, 2013 | |
| c.833G>T | p.Gly278Val | 6 | Coats plus | Bisserbe et al, 2015 | |
| c.841T>C | p.Tyr281His | 6 | Coats plus | Bisserbe et al, 2015 | |
| c.859C>T | p.Arg287* | 6 | Coats plus | Anderson et al,2012 | |
| c.1058delC | p.Ser353Leufs*14 | 6 | cerebroretinal microangiopathy | Polvi et al., 2012 | |
| c.1213delG | p.Asp405MetfsX57 | 8 | Norrie disease, cerebroretinal microangiopathy | Romaniello et al, 2012 | |
| c.1270T>G | p.Cys424Gly | 8 | dyskeratosis congenita | Walne et al, 2013 | |
| c.1389_1390insGTTAGGA | p.Leu464Valfs*66 | 8 | dyskeratosis congenita | Walne et al, 2013 | |
| c.1451A>C | p.His484Pro | 9 | Coats plus | Netravathi et al, 2015 | |
| c.1507G>C | p.Gly503Arg | 9 | Coats plus | Anderson et al, 2012 | |
| c.1994T>G | p.Val665Gly | 12 | cerebroretinal microangiopathy | Polvi et al, 2012 | |
| c.1994T>G | p.Val665Gly | 12 | idiopathic portal hypertension | Wartiovaara-Kautto et al, 2016 | |
| c.2518C>T | p.Arg840Trp | 15 | Coats plus | Anderson et al, 2012 | |
| c.2611G>A | p.Val871Met | 15 | Coats plus | Anderson et al, 2012 | |
| c.2831delC | p.Pro944Leufs*7 | 17 | cerebroretinal microangiopathy | Polvi et al, 2012 | |
| c.2831dupC | p.His945Serfs*56 | 17 | Coats plus | Anderson et al, 2012 | |
| c.2923A>G | p.Arg975Gly | 17 | Coats Plus | Anderson et al, 2012 | |
| c.2923A>G | p.Arg975Gly | 17 | cerebroretinal microangiopathy | Polvi et al, 2012 | |
| c.2954_2956delGTT | p.Cys985del | 18 | Coats plus | Anderson et al, 2012 | |
| c.2954_2956delGTT | p.Cys985del | 18 | cerebroretinal microangiopathy | Polvi et al, 2012 | |
| c.2954_2956delGTT | p.Cys985del | 18 | dyskeratosis congenita | Keller et al, 2012 | |
| c.2954_2956delGTT | p.Cys985del | 18 | dyskeratosis congenita | Walne et al, 2013 | |
| c.2959C>T | p.Arg987Trp | 18 | Coats plus | Anderson et al, 2012 | |
| c.2959C>T | p.Arg987Trp | 18 | dyskeratosis congenita | Walne et al, 2013 | |
| c.3019delC | p.Leu1007Cysfs*62 | 19 | cerebroretinal microangiopathy | Polvi et al, 2012 | |
| c.3425_3426delTCinsAT | p.Leu1142His | 22 | cerebroretinal microangiopathy | Polvi et al, 2012 | |
| c.3583C>T | p.Arg1195* | 23 | cerebroretinal microangiopathy | Polvi et al, 2012 | |
| c.3586_3606del | p.Leu1196_Arg1202del | 23 | Coats plus | Anderson et al, 2012 | |
| c.3586_3606del | p.Leu1196_Arg1202del | 23 | cerebroretinal microangiopathy | Polvi et al, 2012 |
RTEL1
RTEL1 mutations (IPF, DKC and hoyeraal-hreidarsson syndrome)
Numerous mutations causing amino acid substitutions, deletions, and the loss of the entire exon 15 within RTEL1, a telomerase-associated protein, have been connected with human diseases. The RTEL1 protein is a DNA helicase which functions in the protection, stability and elongation of telomeres in humans. RTEL1 interacts with the TRF2, TRF1 and TIN2 proteins in the shelterin complex located at the ends of telomeres to protect telomeres from homologous recombination during DNA replication.
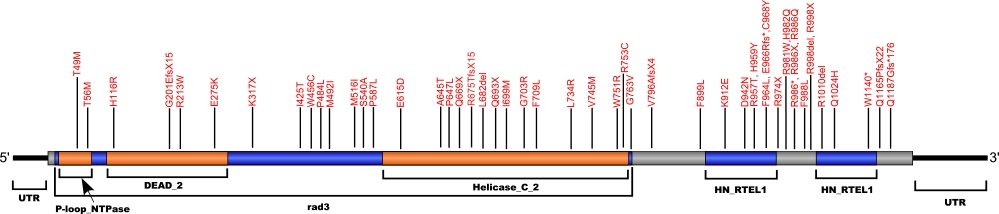
The above schematic shows the larger domains that form the 5042 bp RTEL1 transcript variant 2 gene. The location of some of mutations known to be associated with genetic diseases are shown. Due to the various transcript variants of the RTEL1 gene it is not possible to correctly and accurately show all identified mutations. The black line represents the genomic DNA sequence. Colored boxes indicate translated regions of cDNA and the domains they are located in. The Genbank accession numbers used are NG_033901.1 for the genomic DNA sequence and NM_032957.4 for the cDNA and amino acid sequences. Below is the 1243 amino acid sequence for RTEL1 protein with the various domains colored and labeled. The mutated residues are colored red and the change in amino acids is labeled above.
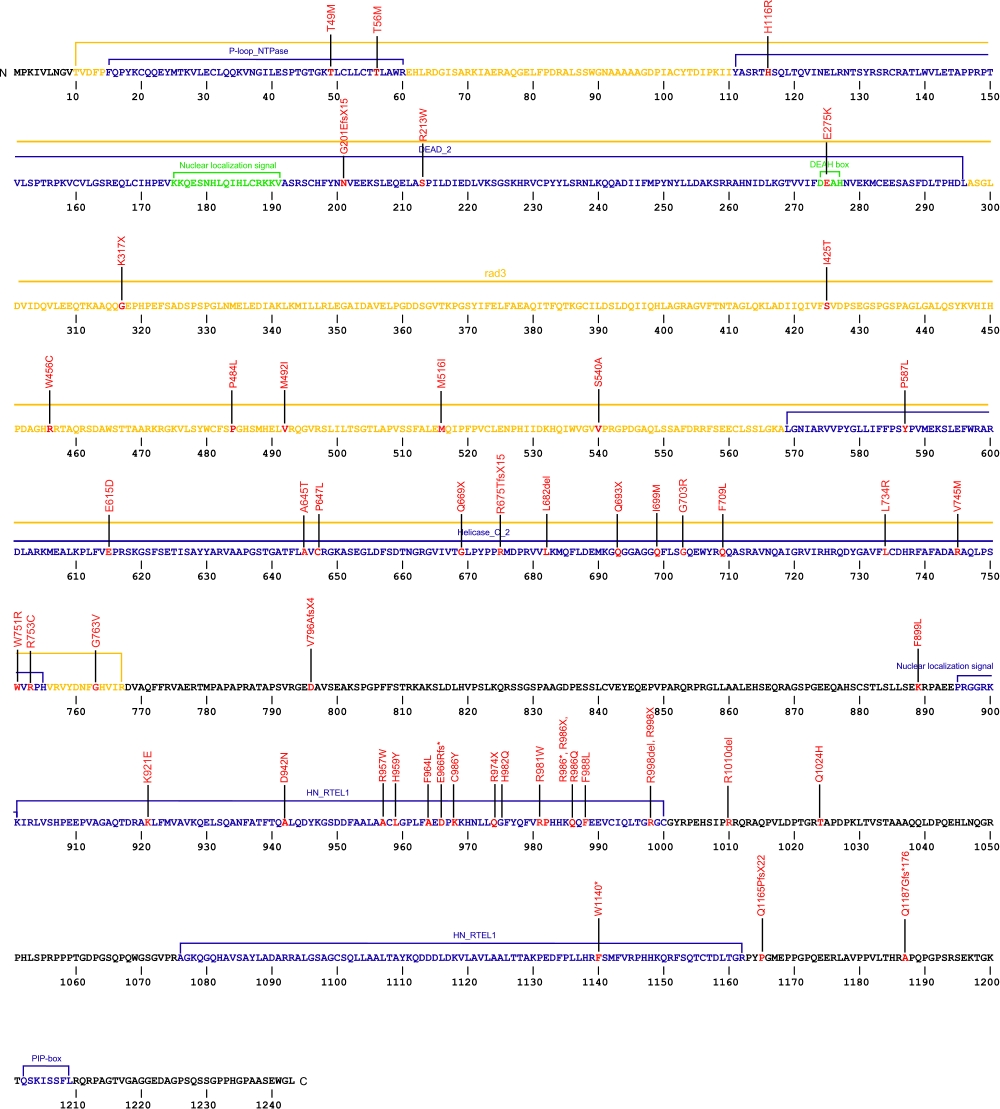
Note: Due to RTEL1 mutations being identified in different isoforms, it is not possible to include all mutations listed below on the same schematic.
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the telomerase-associated protein RTEL1. The cDNA sequence is used for the nucleotide sequence. The mutations are organized by isoform and then by domain from the N-terminus to the C-terminus. Mutations reported without information on the isoform are designated as (?). The NCBI reference sequences used for isoforms 1, 2, and 3 of RTEL1 are, respectively, NP_057518.1, NP_116575.3, and NP_001269938.1.
RTEL1 mutations
| Domains | Isoform | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|---|
| 1 | c.949A>T | p.Lys317X | 11 | hoyeraal-hreidarsson syndrome | Jullien et al, 2016 | |
| c.(?) | p.Ile398_Lys422del | 15 skipped | hoyeraal-hreidarsson syndrome | Jullien et al, 2016 | ||
| c.1476G>T | p.Met492Ile | 17 | hoyeraal-hreidarsson syndrome and myelodysplastic syndrome | Deng et al, 2013 Keel et al, 2016 |
||
| c.2920C>T | p.Arg974X | 30 | hoyeraal-hreidarsson syndrome and familial interstitial pneumonia | Cogan et al, 2015 Deng et al, 2013 |
||
| 2 | c.102+2T>C | n/a | IVS | dyskeratosis congenita | Walne et al, 2013 | |
| c.146C>T | p.Thr49Met | 3 | pulmonary fibrosis | Kannengiesser et al, 2015 Borie et al, 2019 |
||
| c.637C>T | p.Arg213Trp | 8 | pulmonary fibrosis | Kannengiesser et al, 2015 Borie et al, 2019 |
||
| c.823G>A | p.Glu275Lys | dyskeratosis congenita | Walne et al, 2013 | |||
| .1228_1266+39del78, c.1266+3>G | NA | Pulmonary Fibrosis | Borie et al, 2019 | |||
| c.1263+3A>G | p.422_446del | IVS | dyskeratosis congenita | Walne et al, 2013 | ||
| c.1548G>T | p.Met516Ile | hoyeraal-hreidarsson syndrome and dyskeratosis congenita | Fedick et al, 2015 Walne et al, 2013 |
|||
| c.1773G>T | p.Glu615Asp | dyskeratosis congenita and hoyeraal-hreidarsson syndrome | Ballew et al, 2013 | |||
| c.1933G>A | p.Ala645Thr | 22 | dyskeratosis congenita | Ballew et al, 2013 | ||
| c.(?) g.62320919A>AC | p.Arg675ThrfsX15 | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |||
| c.2044_20 46delCTC | p.Leu682del | Pulmonary Fibrosis | Justet et al, 2021 | |||
| c.2097C>G | p.Ile699Met | 24 | hoyeraal-hreidarsson syndrome | Le Guen et al, 2013 | ||
| c.(?) g.623321112G>A | p.Gly703Arg | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |||
| c.2201T>G | p.Leu734Arg | dyskeratosis congenita | Walne et al, 2013 | |||
| c.2214-2A>C | IVS | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |||
| c.2233G>A | p.Val745Met | 25 | hoyeraal-hreidarsson syndrome | Le Guen et al, 2013 | ||
| c.2266-1G>C | NA | Pulmonary Fibrosis | Borie et al, 2019 | |||
| c.2695T>C | p.Arg213Trp | Pulmonary Fibrosis | Borie et al, 2019 Justet et al, 2021 |
|||
| c.(?) g.62321477T>C | p.Trp751Arg | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |||
| c.(?) g.62321483C>T | p.Arg753Cys | idiopathic pulmonary fibrosis | Petrovski et al, 2017 | |||
| c.2869C>T £ | p.Arg957Trp | Pulmonary Fibrosis | Borie et al, 2019 | |||
| c.2869C>T | p.Arg957Trp homozygous | Høyeraal-Hreidarsson | Takedachi et al, 2020 | |||
| c.2288G>T | p.Gly763Val | hoyeraal-hreidarsson syndrome and dyskeratosis congenita | Fedick et al, 2015 Walne et al, 2013 |
|||
| c.2896del | p.Glu966Argfs* | Pulmonary Fibrosis | Borie et al, 2019 | |||
| c.2761A>G | p.Lys921Glu | dyskeratosis congenita | Walne et al, 2013 | |||
| c.2890T>C | p.Phe964Leu | 30 | pulmonary fibrosis | Kannengiesser et al, 2015 Borie et al, 2019 |
||
| c.2903G>A | p.Cys968Tyr | Pulmonary Fibrosis | Justet et al, 2021 | |||
| c.2941C>T | p.Arg981Trp | dyskeratosis congenita and hoyeraal-hreidarsson syndrome | Walne et al, 2013 Fedick et al, 2015 |
|||
| c.2946C>G | p.His982Gln | Pulmonary Fibrosis | Justet et al, 2021 | |||
| c.2956C>T | p.Arg986* | Pulmonary Fibrosis | Borie et al, 2019 Justet et al, 2021 |
|||
| c.2964T>G | p.Phe988Leu | dyskeratosis congenita | Walne et al, 2013 | |||
| c.2992C>T | p.Arg998X | 30 | idiopathic pulmonary fibrosis, dyskeratosis congenita and hoyeraal hreidarsson syndrome | Petrovski et al, 2017 Ballew et al, 2013 |
||
| c.(?) g.62324600C>T | p.Arg1010X | 30 | dyskeratosis congenita, hoyeraal-hreidarsson syndrome and idiopathic pulmonary fibrosis | Petrovski et al, 2017 Ballew et al, 2013 |
||
| c.3420C>A | p.Trp1140* | Pulmonary Fibrosis | Borie et al, 2019 | |||
| c.3493dupC | p.Gln1165ProfsX22 | 33 | pulmonary fibrosis | Kannengiesser et al, 2015 Justet et al, 2021 |
||
| c.3556_3559delAGAC | p.Gln1187Gly fs*176 | Pulmonary Fibrosis | Borie et al, 2019 | |||
| c.3791G>A | p.Arg1264His | hoyeraal-hreidarsson syndrome, natural killer lymphocyte deficiency, myelodysplastic syndrome, dyskeratosis congenita and familial interstitial pneumonia | Fedick et al, 2015 Hanna et al, 2015 Keel et al, 2016 Walne et al, 2013 Cogan et al, 2015 |
|||
| 3 | c.602delG | p.Gly201GlufsX15 | idiopathic pulmonary fibrosis | Stuart et al, 2015 | ||
| c.1135+1G>A | Myelodysplastic syndrome and Acute myeloid leukemia | Schratz et al, 2020 | ||||
| c.1274T>C | p.Ile425Thr | dyskeratosis congenita | Speckmann et al, 2017 | |||
| c.1368G>T | p.Trp456Cys | dyskeratosis congenita | Speckmann et al, 2017 | |||
| c.1451C>T | p.Pro484Leu | idiopathic pulmonary fibrosis | Stuart et al, 2015 | |||
| c.1760C>T | p.Pro587Leu | Myelodysplastic syndrome and Acute myeloid leukemia | Schratz et al, 2020 | |||
| c.1940C>T | p.Pro647Leu | idiopathic pulmonary fibrosis | Stuart et al, 2013 | |||
| c.2005C>T | p.Gln693X | idiopathic pulmonary fibrosis | Stuart et al, 2015 | |||
| c.2387delT | p.Val796AlafsX4 | dyskeratosis congenita | Speckmann et al, 2017 | |||
| c.2652+5G>C | p.Pro884_Gln885ins53X13 | IVS28 | dyskeratosis congenita | Speckmann et al, 2017 | ||
| c.2869C>T | p.Arg957Trp | hoyeraal-hreidarsson syndrome | Touzot et al, 2016 | |||
| c.2892T>G | p.Phe964Leu | 30 | hoyeraal-hreidarsson syndrome | Touzot et al, 2016 | ||
| c.3371A>C | p.His1124Pro | idiopahtic pulmonary fibrosis | Stuart et al, 2015 | |||
| c.3730T>C | p.Cys1244Arg | 34B | hoyeraal-hreidarsson syndrome | Le Guen et al, 2013 | ||
| c.3730delTG | p.Cys1244ProfsX17 | dyskeratosis congenita | Speckmann et al, 2017 | |||
| c.3802T>C | p.Cys1268Arg | hoyeraal-hreidarsson syndrome | Touzot et al, 2016 | |||
| c.3880G>T | p.Val1294Phe | hoyeraal-hreidarsson syndrome | Touzot et al, 2016 | |||
| (?) | c.102+1G>A | n/a | IVS | cryptic dyskeratosis congenita | Yamaguchi et al, 2015 | |
| c.(?) | p.Thr56Met | myelodysplastic syndrome | Keel et al, 2016 | |||
| c.(?) | p.His116Arg | myelodysplastic syndrome | Keel et al, 2016 | |||
| c.958+1G>GT | n/a | IVS10 | familial interstitial pneumonia | Cogan et al, 2015 | ||
| c.1482-1G>A | n/a | IVS | afmilial interstitial pneumonia | Cogan et al, 2015 | ||
| c.1618T>G | p.Ser540Ala | familial interstitial pneumonia | Cogan et al, 2015 | |||
| c.(?) | p.Gln669X | chronic hypersensitivity pneumonitis | Newton et al, 2016 | |||
| c.(?) | p.Phe709Leu | cryptic dyskeratosis congenita | Yamaguchi et al, 2015 | |||
| c.2219_2227del | p.740_742del | familial interstitial pneumonia | Cogan et al, 2015 | |||
| c.2413+1G>C | n/a | IVS25 | familial interstitial pneumonia | Cogan et al, 2015 | ||
| c.2695T>C | p.Phe899Leu | idiopathic pulmonary fibrosis | Juge et al, 2017 | |||
| c.2824G>A | p.Asp942Asn | idiopathic pulmonary fibrosis | Juge et al, 2017 | |||
| c.2875C>T | p.His959Tyr | idiopathic pulmonary fibrosis | Juge et al, 2017 | |||
| c.2956C>T | p.Arg986X | hoyeraal-hreidarsson syndrome | Moriya et al, 2016 | |||
| c.2957G>A | p.Arg986Gln | familial interstitial pneumonia | Cogan et al, 2015 | |||
| c.3126A>C | p.Gln1024His | hoyeraal-hreidarsson syndrome | Mariya et al, 2016 |
NAF1
NAF1 mutations
Currently, there are two frame shift mutataions that have been identified in the NAF1 protein. NAF1 is part of an H/ACA motif-binding complex, which consists of the proteins dyskerin, NHP2, NOP10, and NAF1. The assembly of this complex with hTR transcript begins the biogenisis of the human telomerase holoenzyme.
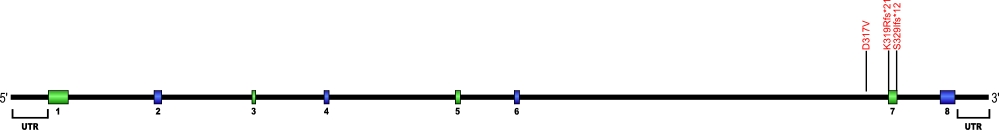
The above structural organizational scheme for the eight exons that form the NAF1 gene has indicated the location of mutations known to cause human diseases. The black line represents the genomic DNA sequence with the untranslated regions (UTR) labeled. Colored boxes indicate exons 1-8. The Genbank accession number used was NM_138386.2 for the cDNA and amino acid sequences. Below is the 494 amino acid sequence for the NAF1 protein with the 8 exons numbered and labeled. The mutated residues are colored red and the change in amino acids is labeled above.
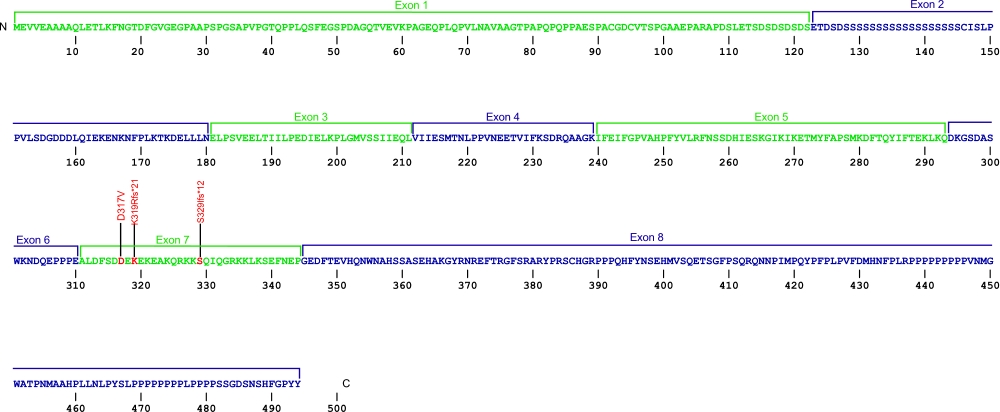
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the NAF1 protein. Both the cDNA sequence and genomic position are used for the nucleotide sequence. The mutations are organized by domain from the N-terminus to the C-terminus.
NAF1 mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| c.956_957delAA | p.Lys319Argfs*21 | 7 | Idiopathic Pulonary Fibrosis | Stanley et al, 2016 | |
| c.984insA | p.Ser329Ilefs*12 | 7 | Pulmonary Fibrosis-Emphysema | Stanley et al, 2016 | |
| c.950A>T | p.Asp317Val | 7 | Myelodysplastic syndrome / Acute myeloid leukemia | Schratz et al, 2021 |
ZCCHC8
ZCCHC8 mutations
Currently, there is one substitution mutataion that has been identified in the ZCCHC8 protein. ZCCHC8 is a component of the nuclear exosome targeting complex (NEXT), involved in the processing of intronless RNAs, such as telomerase RNA, a special class of replication dependent histones, and RNAs that encode cilia protein components.
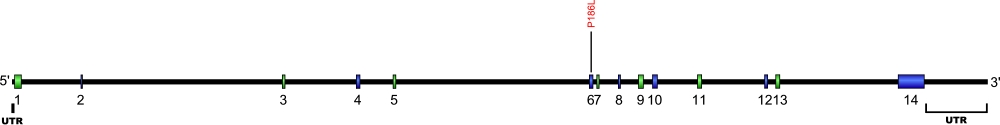
The above structural organizational scheme for the fourteen exons that form the ZCCHC8 gene have indicated the location of the mutation known to cause human disease. The black line represents the genomic DNA sequence with the untranslated regions (UTR) labeled. Colored boxes indicate amino acid coding sequences 1-14. The Genbank accession number used was NM_017612.5 for the cDNA and amino acid sequences. Below is the 707 amino acid sequence for the ZCCHC8 protein with the 14 exons numbered and labeled. The mutated residues are colored red and the change in amino acids is labeled above.
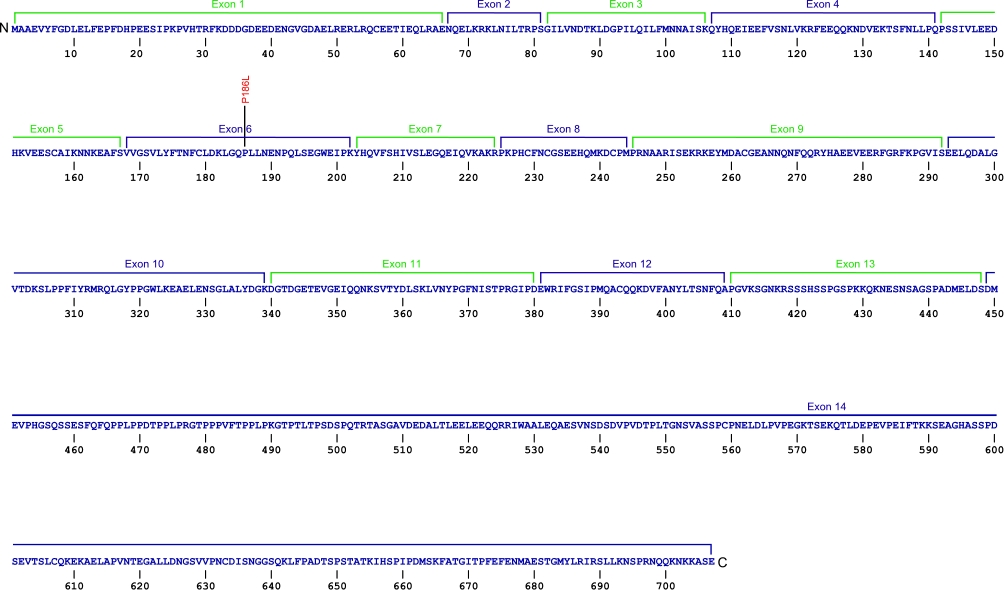
Below is a description of the locations within the nucleotide and protein sequence, the amino acid substitutions, the characteristic clinical presentations, and original literature citations that are linked to the published online journal for known mutations within the ZCCHC8 protein. Both the cDNA sequence and genomic position are used for the nucleotide sequence. The mutations are organized by domain from the N-terminus to the C-terminus.
ZCCHC8 mutations
| Domains | Mutation | AA substitution | Exon | Presentation | References |
|---|---|---|---|---|---|
| c.C557T | p.Pro186Leu | 6 | Familial Idiopathic Pulmonary Fibrosis | Gable et al, 2019 |